
NCBI(National Center for Biotechnology Information)が提供するSequence Read Archive(SRA)から取得したシーケンスデータでSingle cell RNA-seq解析したいと思いませんか?
この記事は、SRAを10x GenomicsのCell Rangerでデータ処理する方法を解説します。
データの取得から、FASTQファイルのダウンロード、そしてFASTQファイルの命名規則に従った名前変更までの方法を解説します。
この記事を理解することで、公開されているシーケンスデータを効率的に再利用することができますぜひ挑戦してみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB, cellranger 7.1.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
Cell Rangerとは?
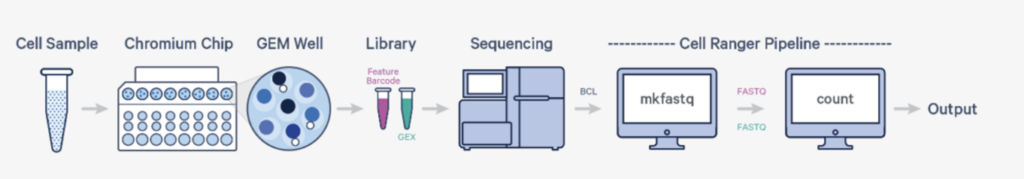
Cell Rangerは10x Genomics社が開発したソフトウェアパッケージで、シングルセルRNAシーケンシング(scRNA-seq)データの前処理と解析を行うためのツールです。10x GenomicsのChromiumシステムを用いたシングルセルRNA-seq実験から得られるデータの解析に特化しています。下記の図でいうとsequencingした後のデータ処理を行う部分を指しています。Cell Rangerから出力されたOutputはデータの次元削減(t-SNEやPCA)、クラスタリング、差異発現解析など下流の解析へと用いられていきます。
前回の記事でcell rangerの基本的な使い方は解説しました。
まだ確認されていない方はそちらの方を先に取り組むことでcell rangerの使い方を勉強できるようになっています。チェックしてみてください。
NCBIのSRAデータをCell Ranger用に準備する方法
それでは実際にNCBIのSRAデータをCell Ranger用に準備する方法を解説していきます。
NCBI SRAからfastqファイルをダウンロードする
fastq-dump コマンドを使うのでsra-toolkitをインストールしておいてください。
Macはこちら
Windowsはこちら (Ubuntuをインストールしてない方は、Ubuntuをインストールから始めてください)
最初に、--split-files引数を使用してNCBIのfastq-dumpユーティリティでFASTQファイルを取得します。コマンドは次のようになります
fastq-dump --split-files --gzip SRR6334436
すると以下の2つのfastqファイルがダウンロードされるかと思います。
SRR6334436_1.fastq.gz
SRR6334436_2.fastq.gz
こちらはなにか判別するためにSRR6334436のMetadataをまずは見てみましょう。
注目するのは、L=26, L=98という数字です。Read1 とRead2のリード長を表しています。
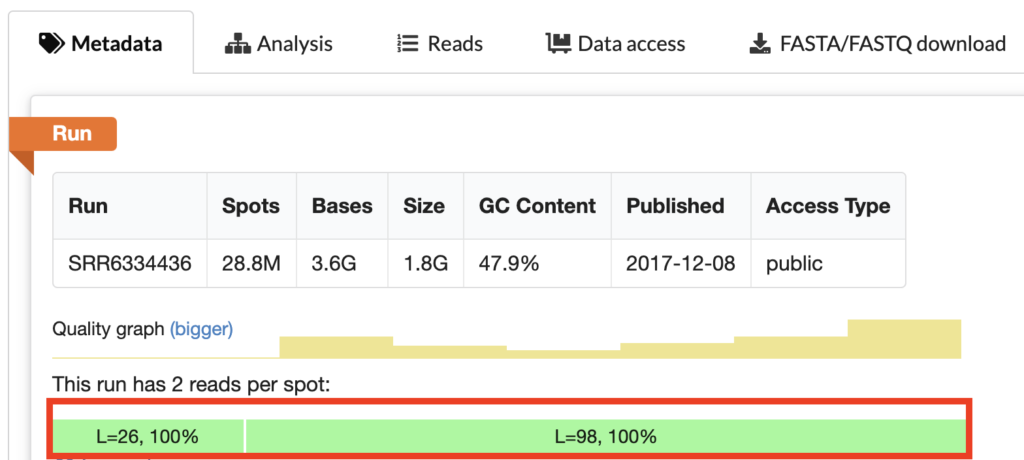
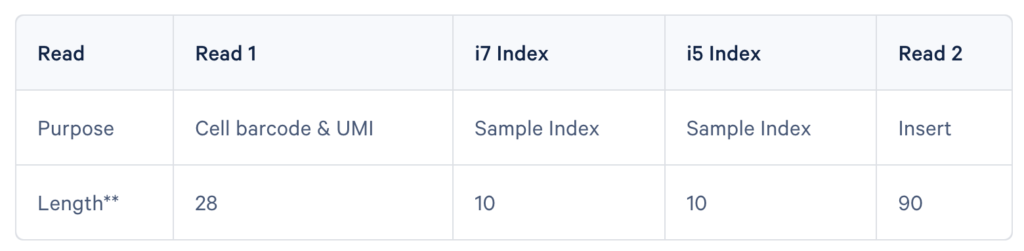
Read1 は Cell barcode & UMIのデータであり、Read2はInsertを表しています。それぞれのLengthは28, 90であり、このデータから、SRR6334436_1.fastqはRead1、SRR6334436_2.fastqはRead2であると推測することができます。
通常、シーケンスデータのサンプルは2つのリード(Read 1とRead 2)を持っていますが、場合によっては3つまたは4つのリードを持つことがあります。これは、インデックスリードが含まれている場合によく見られます。
例として、SRR9291388からFASTQファイルを取得する場合を考えてみましょう。
コマンドは以下の通りです。
fastq-dump --split-files --gzip SRR9291388
このコマンドを実行すると、出力は3つのFASTQファイルになります。
SRR9291388_1.fastq.gz
SRR9291388_2.fastq.gz
SRR9291388_3.fastq.gz
この結果は、SRR9291388のメタデータに報告されている情報と一致しています。すなわち、このランは1スポットあたり3つのリードを持っています。
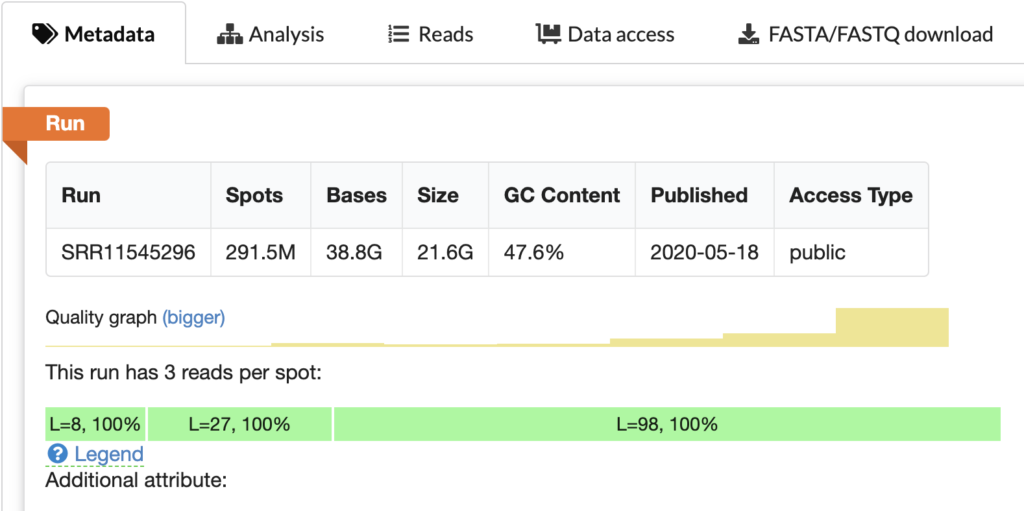
このMetadataから以下のようにそれぞれのファイルが割り当てることができます。
SRR9291388_1.fastq :Read 1SRR9291388_2.fastq :Read 2SRR9291388_3.fastq :Index
ファイル名を変更する
Cell Rangerはbcl2fastqのファイル命名規則に従ったFASTQファイル名が必要です。bcl2fastqのファイル命名規則とは以下のような名前になります。
[サンプル名]_S1_L00[レーン番号]_[リードタイプ]_001.fastq.gz
各部分を解説します。
- [Sample Name]: この部分はサンプルの名前またはIDを表します。これによって、どのサンプルに対応するデータであるかが識別されます。
- S1: これはサンプルのシリアル番号を表します。複数のサンプルがある場合、
S2,S3,S4…といったように番号が増えていきます。 - L00[Lane Number]: この部分はフローセル内のレーン番号を示します。Illuminaのシーケンサーは多くの場合、複数のレーンを持っており、この番号によってそれらが区別されます。例えば、
L001,L002など。 - [Read Type]: この部分はリードの種類を示します。通常、
R1はフォワードリード(Read 1)を、R2はリバースリード(Read 2)を表します。インデックスリードは通常I1(Index 1)、I2(Index 2)などと表されます。ここで、リードタイプは以下のいずれかです。- I1: サンプルインデックスリード(オプション)
- I2: サンプルインデックスリード(オプション)
- R1: Read 1(必須)
- R2: Read 2(必須)
- 001: この部分はセグメント番号を示します。通常は
001とされますが、特定のケースで複数のセグメントが必要な場合、この番号が増えていくことがあります。
この規則に基づいて、SRR6334436_1.fastq.gzとSRR6334436_2.fastq.gzのファイル名を変更すると、以下のようになります。
- Read 1:
SRR6334436_S1_L001_R1_001.fastq.gz - Read 2:
SRR6334436_S1_L001_R2_001.fastq.gz
SRR9291388_1.fastq, SRR9291388_2.fastq, SRR9291388_3.fastqのファイル名を変更すると、以下のようになります。
- Read 1:
SRR9291388_S1_L001_R1_001.fastq.gz - Read 2:
SRR9291388_S1_L001_R2_001.fastq.gz - Index 1:
SRR9291388_S1_L001_I1_001.fastq.gz
ダウンロードしたファイルを実行する
では実際にSRR9291388_1.fastq, SRR9291388_2.fastq, SRR9291388_3.fastqを使ってCell Rangerを実行してみましょう。
Cell rangerを実行するにはyardフォルダを作るでしたね。忘れてた方は、前回の記事を復習しましょう。
私は、yardフォルダの中にSRR9291388フォルダとSRR9291388_resultフォルダ を作りました。SRR9291388フォルダの中に、名前変更済みのファイルを入れます。
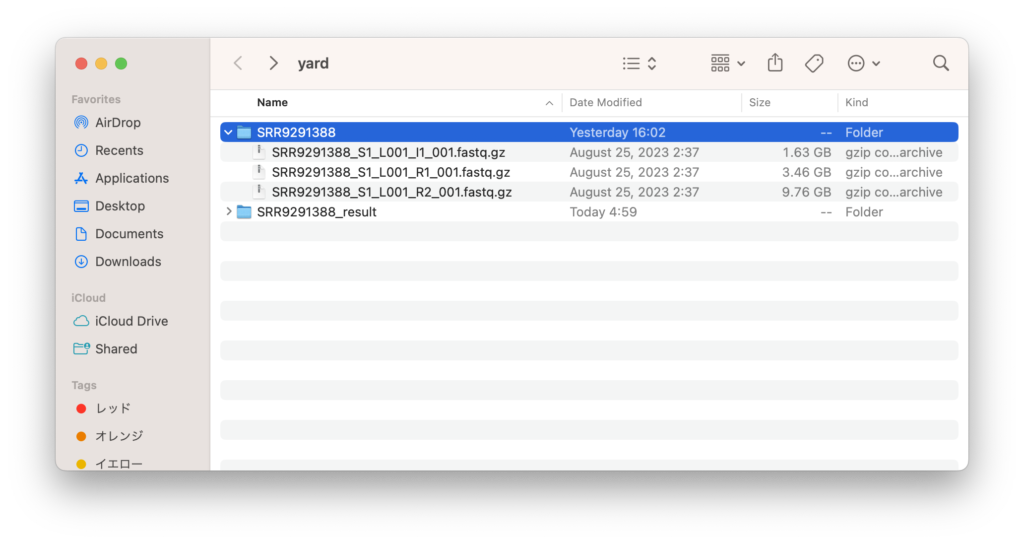
フォルダの準備ができたら下記コマンドを実行してみましょう。
cellranger count --id=SRR9291388_result --fastqs=/yard/SRR9291388 --sample=SRR9291388 --transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A
各オプションの解説は以下のとおりです。
-id=SRR9291388_result: このオプションで指定されたIDは、解析結果の出力ディレクトリ名に使用されます。SRR9291388_resultという名前のディレクトリが作成され、その中に解析結果が保存されます。-fastqs=/yard/SRR9291388: このオプションで指定されたパスは、入力として使用するFASTQファイルが保存されているディレクトリを指します。/yard/SRR9291388ディレクトリ内のFASTQファイルが解析に使用されます。-sample=SRR9291388: このオプションで指定されたサンプル名は、FASTQファイル内で解析するサンプルを識別するために使用されます。この例では、SRR9291388という名前のサンプルが解析されます。-transcriptome=/yard/desktop/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A: このオプションで指定されたパスは、参照トランスクリプトームデータが保存されているディレクトリを指します。この例では、/yard/run_cellranger_count/refdata-gex-GRCh38-2020-Aディレクトリ内の参照トランスクリプトーム(GRCh38バージョン2020-A)が使用されます。
実行結果
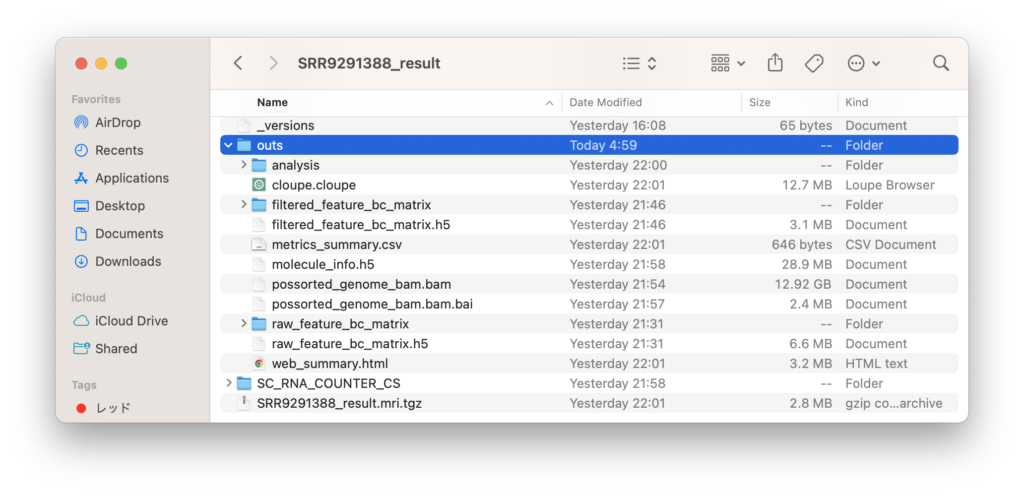
SRR9291388_resultフォルダの中にファイルが出力されていたら成功です!outsの中を見てみると、ほしいファイルができていることがわかると思います。
最後に
いかがだったでしょうか。NCBI SRAのような公共データベースにあるscRNA-seqデータを解析するイメージがついたのではないでしょうか?こちらはNCBI SRA以外のファイルでも適用できます。ぜひ挑戦してみてください!

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

