
RNA-seqを用いた全ゲノム解析のやり方を知りたいと思いませんか?
RNA−seq解析はドライ系のバイオインフォマティシャンの人だけがやるんでしょ..?と考えている実験系の人は多いかもしれませんが、近年はRNA−seq解析の敷居が下がり、実験系の人でも取り入れることが可能になってきております。
前回はMac向けのikraの使い方を書きましたが、実際の多くの研究者はWindowsを使っていると思いますので、今回はLinuxのディストリビューションであるUbuntuを入れることで、Windowsでもikraを動かす方法を紹介したいと思います。
デバイス名 K-DELL
プロセッサ 12th Gen Intel(R) Core(TM) i5-1240P 1.70 GHz
実装 RAM 8.00 GB
システムの種類 64 ビット オペレーティング システム、x64 ベース プロセッサ

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
ikraとは?
ikraはRNA-seq解析を完全自動化したツール
ikraはRNA-seqの生データから、発現量データをTSVファイルとして出力する解析パイプラインです。
RNA-seq解析を行うには複数のツールを組み合わせて段階的に解析していく必要がありますが、ikraではすべてのツールをパイプラインとしてつなぐことで、実行コマンド一つで自動的に各ツールが実行されていく仕組みになっています。
そのため、RNA-seq解析初心者が各ツールの使い方がわからず挫折する、、といった事態を避ける事ができます。
またikraで使用できるRNA-seqデータの生物種はhumanかmouseに限られていますので、ご注意ください。
ikraから出力されるファイル群
ikraから出力されるファイルはクオリティチェックファイル(fast.qc)など複数ありますが、遺伝子発現の定量結果はoutput.tsvに出力されます。今回はこのoutput.tsvを出力するところまでを解説します。
Windowsでikraを動かす上であると良いPCスペック
最低限以下のスペックのPCがあると良いです。
- 最大CPUコア:8
- 最大メモリ:16GB
- デスクトップ型orノート型:ノート型でも大丈夫
- ストレージ:500GB
この中だとメモリの増設難しいため一番重要となってきます。
バイオインフォ系の処理は仮想化環境を作るDockerと呼ばれるソフトを同時に使うことが多く、Dockerはメモリを大きく使うため、8GB程度だと他に何も操作できなくなってしまう状態になってしまいます(解析回しながらパワポ作成、などは重すぎて無理)最低でも16GBがおすすめです。
ちなみにストレージに関しては、外付けHDDなどで拡張できるので、この中で行ったら優先順位を下げて良いです。
Windowsでikraを扱う方法
ではこっからは実際にWindowsでikraを動かすための準備をしていきます。
流れとしては、Ubuntuインストール→sra-toolkitのインストール→Docker Desktopのインストール→ikraを使う準備→ikraへ読み込ませるデータ準備→実際に実行する、になります。
Ubuntuインストール
Linux 用 Windows サブシステムを有効にしていきます。
- PowerShellを「管理者として実行」する
- 下記コマンドで、LinuxのディストリビューションであるUbuntuをインストール
wsl --install -d Ubuntu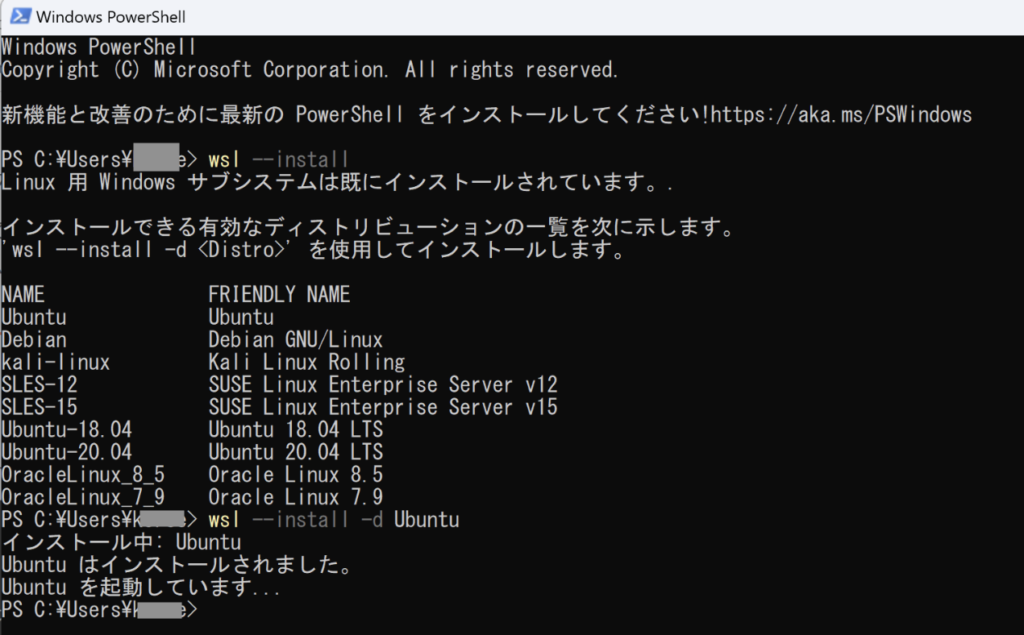
インストールが完了してUbuntuをクリックしてみると下記のようなエラーになり起動できないかと思います。
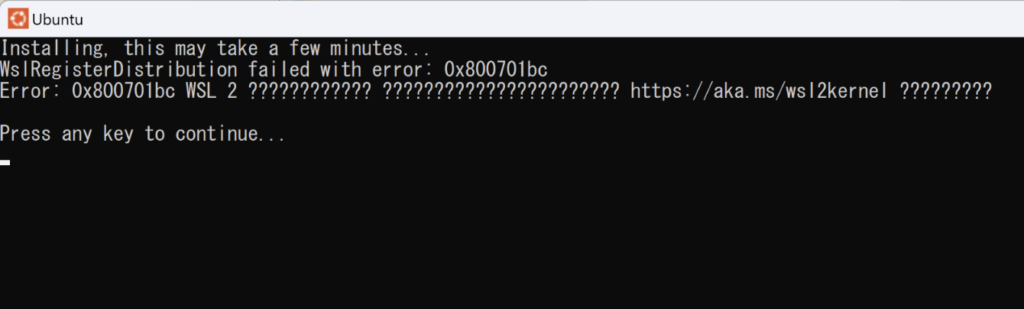
これは、Linux カーネル更新プログラム パッケージをダウンロードする必要あります。
Microsoftの以前のバージョンの WSL の手動インストール手順(下記の画像)のところから最新版のパッケージをダウンロードすることで解決します。
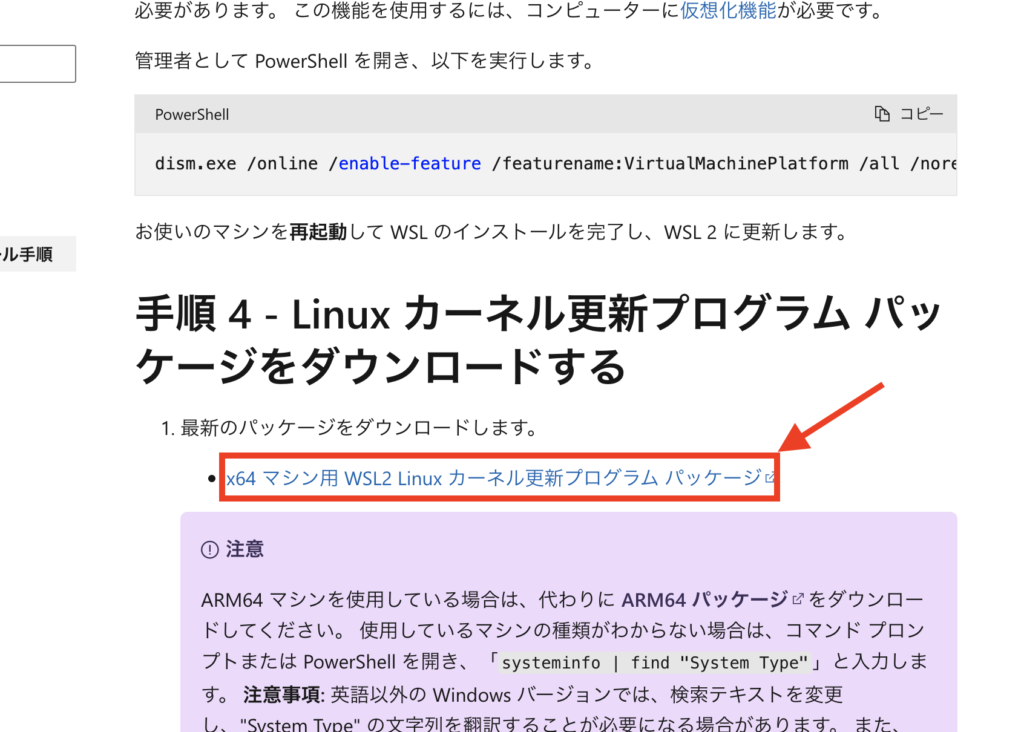
最新版のパッケージをダウンロードしたらインストールを進めてください。
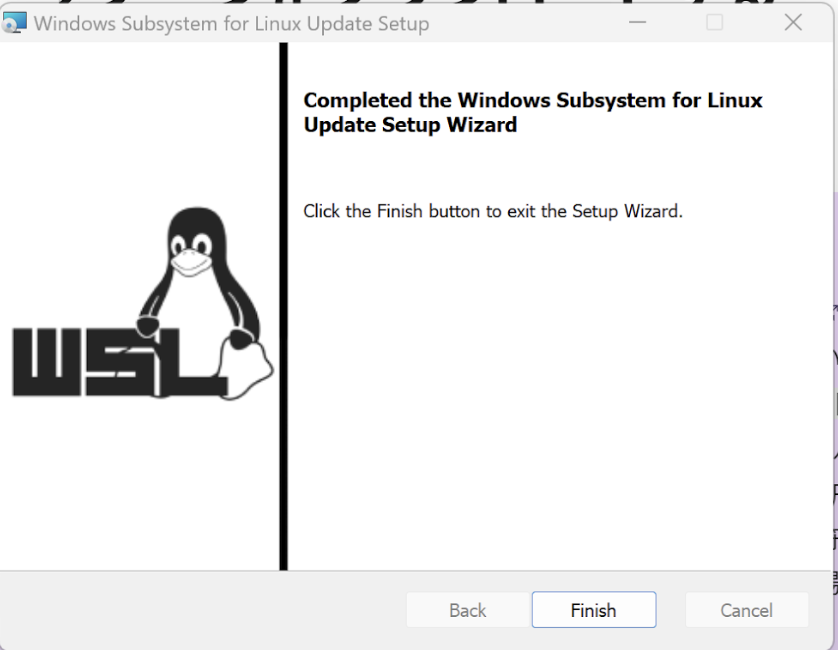
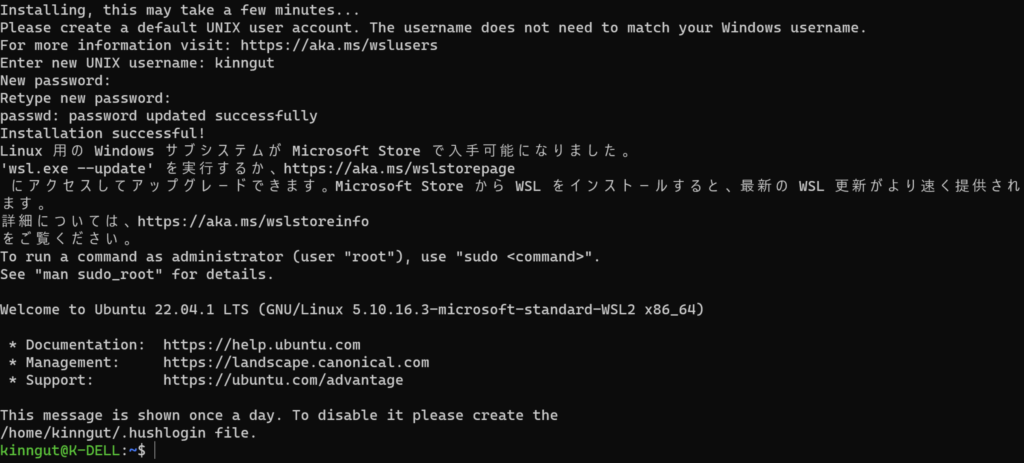
これでUbuntuが起動できるようになります。UNIX usernameとpasswordを決めて起動してください。
sra-toolkitsのインストール
次にローカルにsra-toolkitをダウンロードしていきます。デスクトップに移動しますが、自分の場合は以下のパスで移動できました。[ユーザー名]は適宜自身のものに適宜変更してください。
cd /mnt/c/Users/[ユーザー名]/desktopsra-toolkitsをインストールしていきます。Ubuntu LTS 18.04以前の方は、updateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得してから、
$ sudo apt update -y以下のコマンドでインストールすることができます。
$ sudo apt install sra-toolkitUbuntu LTS 18.04以降のバージョンの方は、古いファイルをインストールすることで使用できます。
$ sudo apt update -yupdateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得しておきます。
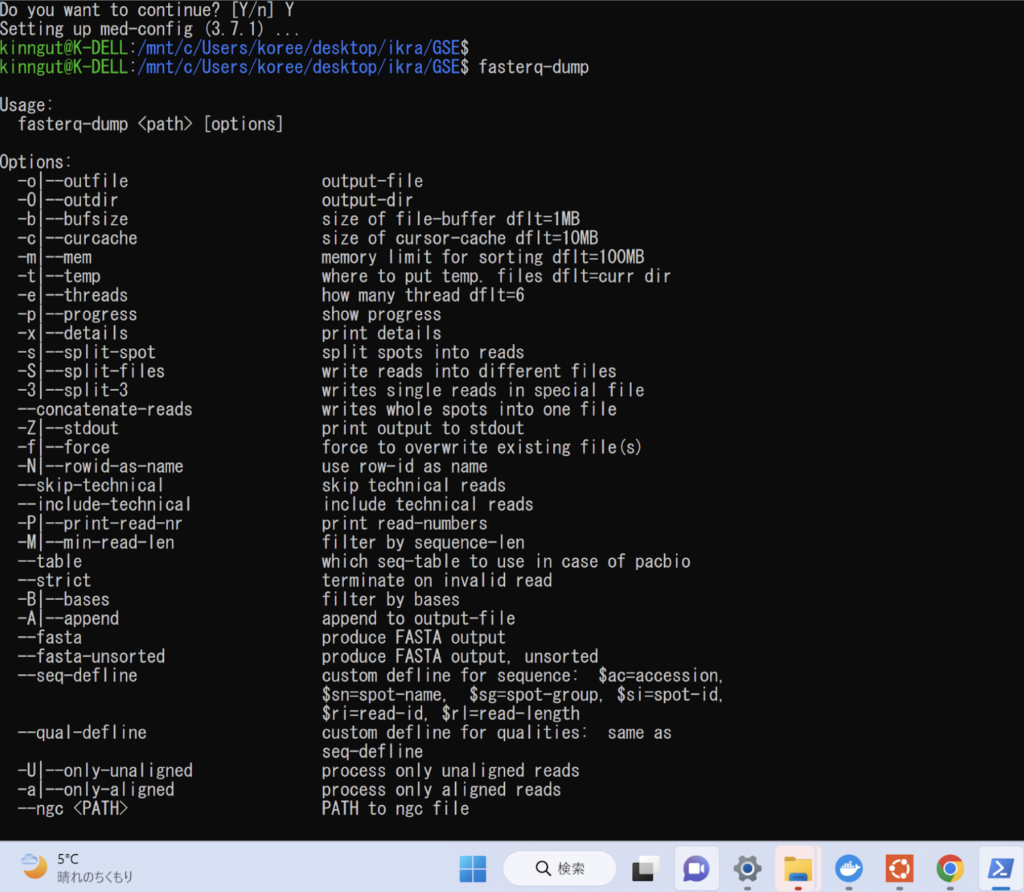
無事installができたらfasterq-dumpコマンドが通るか試してみましょう。以下のような画面になったら正しくインストールできています。
Docker Desktopのインストール
ikraにはDockerを用いるのでDockerをインストールしてください。(ダウンロードに5分程かかります。)
• Windows版 https://docs.docker.com/desktop/windows/install/
インストールが終わったら、Dockerをクリックして起動しておいてください。
ikraを使う準備
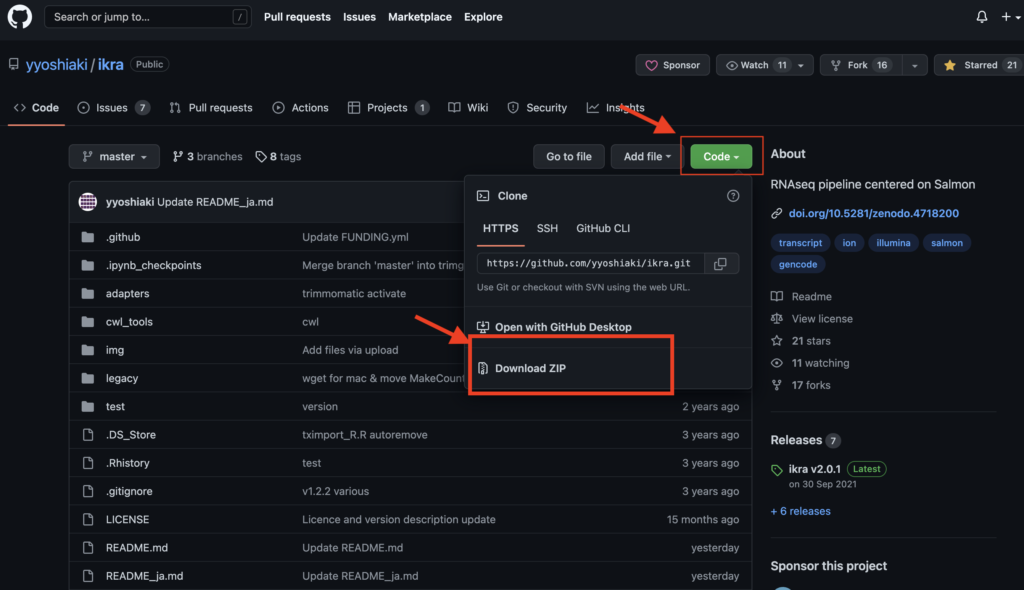
下記URLのGithubより「Code」→「Download ZIP」へ進みzipファイルをダウンロードし、zipファイルを解凍して中身を取り出します。zipファイルはデスクトップで解凍しておいてください。
•https://github.com/yyoshiaki/ikra
ikraへ読み込ませるデータ準備
解析したいSRAデータを探すの主に以下3つの場所があります。
- DBCLS https://sra.dbcls.jp/
- NCBI SRA https://www.ncbi.nlm.nih.gov/sra/
- Array Express https://www.ebi.ac.uk/arrayexpress/
今回はNCBI SRAを用いてデータを探索します。検索バーに探索したいキーワードを入力してください。
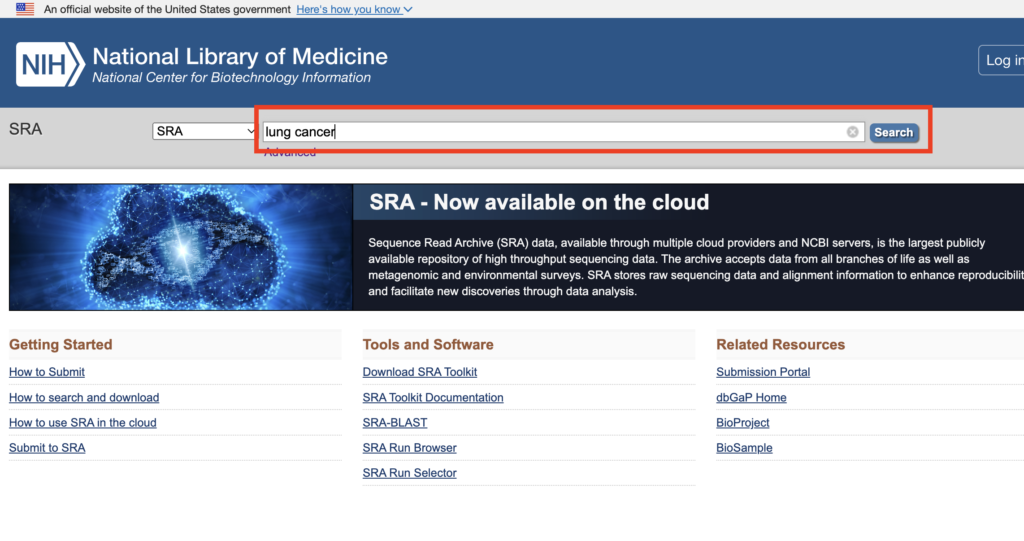
検索結果が出てきます。解析したいデータをクリックしてください。
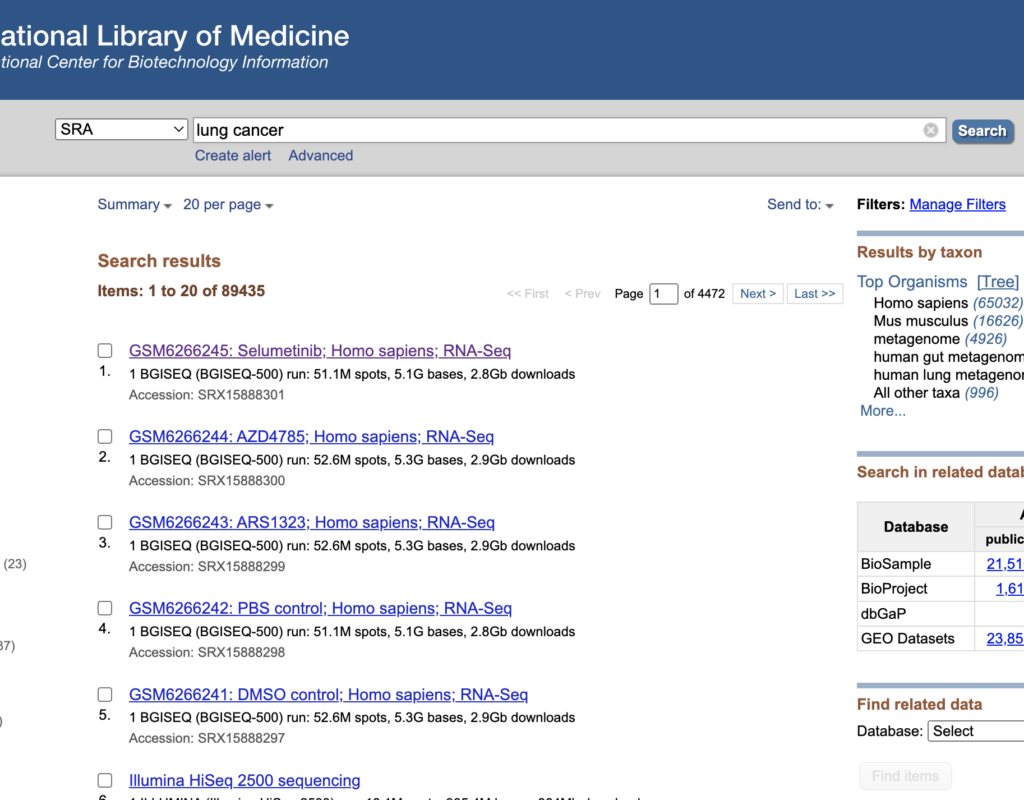
下にスクロールするとSRR~から始まる番号がありますので、こちらをクリックしてください。
SRR番号は解析に用いるので、覚えておいてください。
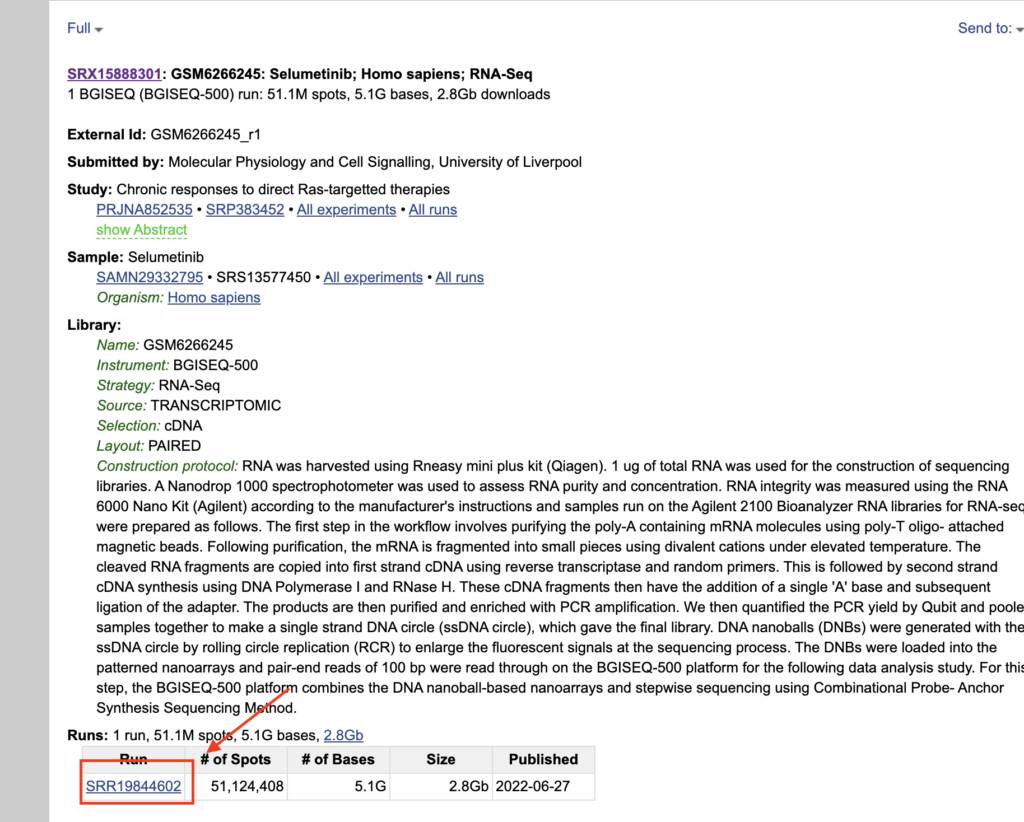
レイアウトがシングルエンドかペアエンドかを確認します。PAIREDになっていたらペアエンド、SINGLEになっていたらシングルエンドです。
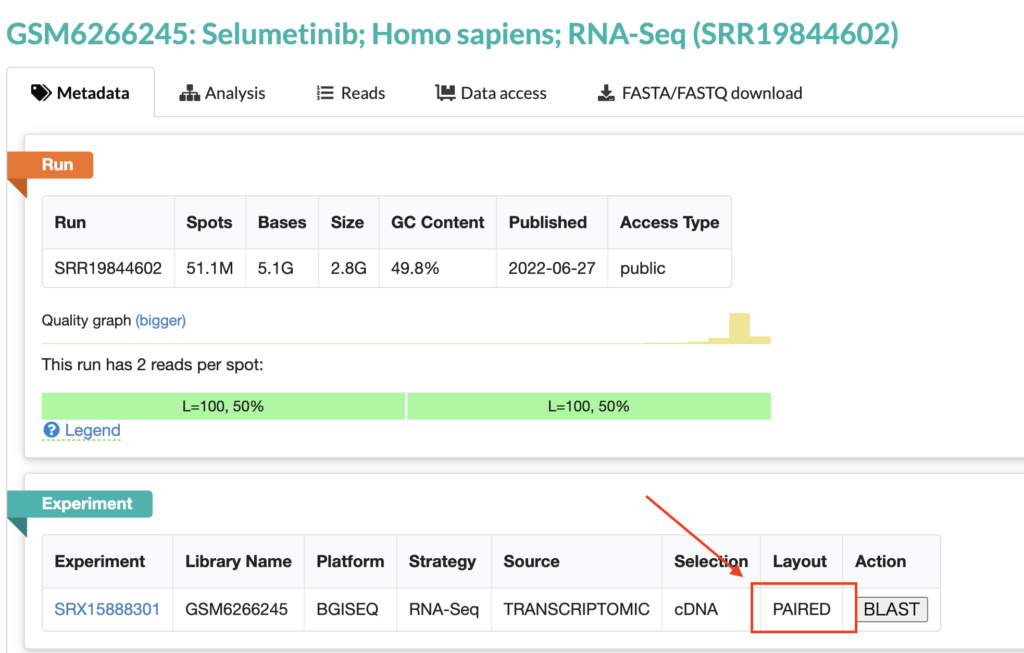
次にikraに読み込ませるcsvファイルを作成していきます。以下の要素が必要です。name, SRR, Layoutという見出しを一行目に記載します。すべてコンマ(,)具切りで書きましょう。
csvファイル名をここでは〇〇.csvと表記していますが、ご自身で好きに命名したファイル名をご使用ください。
- name・・好きな名前。何でも良い
- SRR・・SRR番号
- Layout・・ペアエンドかシングルエンドかの情報。ペアエンドは「PE」、シングルエンドは「SE」と表示します。
name,SRR,Layout
lung_cancer,SRR19844602,PECSVファイル用意ができたら、ikra.shファイルがあるディレクトリと同一のところに、csvファイルをおいてください。
実際にコードを実行してみよう
powershellからikra.shとcsvファイルがあるところに移動し、コマンドを叩いていきます。
$ bash ikra.sh 〇〇.csv human (or mouse)最初は、Dockerimageのダウンロードで5~10分ほど時間がかかります。
fasterq-dumpが走り出したらほぼ成功です。後は4~10時間ほど待てばoutput.tsvファイルができているかと思います。
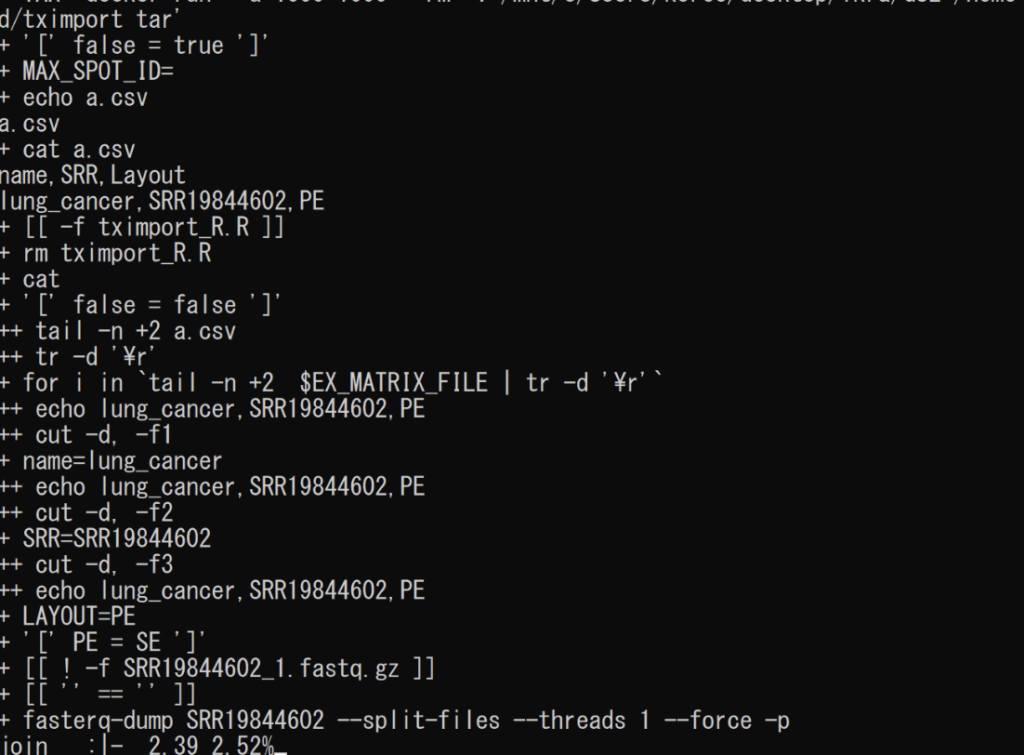
最後に
いかがだったでしょうか。これでWindowsの人でも問題なくikraが動かせる様になるかと思います。研究の合間時間にぜひともチャレンジしてみてください。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます


大変わかりやすく記事を作成していただきありがとうございます。
この記事に沿ってikraを使える環境を構築しています。
一点分からない点がございましたので質問させていただきます。
NCBI SRAからikraに読み込ませるCSVファイルを作成するステップで下記のコードを示していただいております。
name,SRR,Layout
lung_cancer,SRR19844602,PE
こちらは、どちらに入力すればよいかご教師頂けると幸いです。
xihciat様
ご質問いただきありがとうございます。
name,SRR,Layout
lung_cancer,SRR19844602,PE
こちらのコードは作成いただくcsvファイルの中に記載してください。
また作成いただくcsvファイルの名前はご自身で好きに命名してください。
例えば、 `ikra_read.csv` というような名前でcsvファイルを作成します。
csvファイルの作成はWindowsであればメモ帳を使うか、もしくはVS codeなどのエディターを使用すれば作成できます。
補足になりますが、csvファイルはコンマ区切りデータが格納されたファイルを意味します。コンマで区切られたデータが格納されているだけの、ただのテキストファイルです。
マイクロソフト製のExcelで開くことができるので、csvファイルはスプレッドシートであると思ってしまいやすいですが、テキストファイルです。
ご質問いただき、ありがとうございました。
よりわかりやすい記事を執筆していく努力を続けますので、引き続きLabCodeをよろしくお願いいたします。
CSVファイルの作成方法をご教示頂きありがとうございました。
Poweshellに
cd /mnt/c/Users/[ユーザー名]/Desktop/ikra-master
と入力することで、ikraのディレクトリに移動(?)し、
つづいて、Poweshellに
$ bash ikra.sh ikra_read.csv human
と入力することで、無事に実行されました。
ありがとうございます。
度々失礼します。
本記事では、「SRR19844602」という単一のファイルからoutput.tsvを作成する方法をお示していただいております。
複数のFASTQファイルからikraを介して一つのoutput.tsvを作成することは可能でしょうか?
xihciat様
ご質問ありがとうございます。
複数のFASTQファイルからikraを介して一つのoutput.tsvを作成することは可能です。
csvファイルを作成する際に、改行して解析したいサンプルを追加してください。
■CSV例
name,SRR,Layout
lung_cancer_1,SRR19844602,PE
lung_cancer_2,SRR19844603,PE
.
.
.
そうすると以下のようなoutput.tsvの構造が得られると思います。
■output.tsv(出力結果)
lung_cancer_1 lung_cancer_2 lung_cancer_3 lung_cancer_4 lung_cancer_5 lung_cancer_6 lung_cancer_7 lung_cancer_8
A1BG 58.8390292357933 53.8564455187622 56.9019080835063 61.9128730155385 1.90754019283661 1.20581054654057 0 1.62240424300652
A1CF 825.917071200574 755.290297617765 1567.395099937 1797.62403955078 138.401082860673 174.511601832918 170.599951304134 282.572131240043
A2M 9212.66712794542 7854.65176311213 30505.7980285529 32618.8032913106 2.40186074559596 1.50536015691493 1.20822995249316 2.47331488789058
A2ML1 0 0.404832133361214 0 0.416178229112331 0 0 0 1.56261104097179
A3GALT2 0 0 0 0 0 0 0 0
LabCode 様
このたび,記事(公共データを用いたRNA-seq解析)を購入させていただきました.
一点質問させていただいてもよろしいでしょうか.
name,SRR,Layout
lung_cancer,SRR19844602,PE
早速,私も記事で使用されている上記のデータセットを試しに解析してみたのですが,
記事の中に示していただいている実行結果(output.tsv)と私の試した実行結果(output.tsv)では大きく異なった数値を出力してきました.
記事に示していただいてる実行結果(lung_cancer)のA1BG:58.8390292357933・・・のデータは上記のデータセットと同一のものでしょうか?
解析時は特にエラーがあった様子もなかったので,原因が分らずにいます.
考えられる点がありましたらご教授いただければ幸いです.
TM様
ご質問ありがとうございます。
大変申し訳ありませんが、記事で用いているSRR1984460とスクリーンショットで示されているサンプル結果は別のものとなっておりました。
誤解を招いてしまい申し訳ありませんが、別のものとご認識いただけますと幸いです。
よろしくお願い致します。