
scRNA-seqデータ解析である程度の可視化はできたけど、各クラスターに特徴的な遺伝子発現はなんだろうと気になりませんか。
この記事ではscRNA-seqデータのマーカー遺伝子の発現を確認していきます。
マーカー遺伝子の発現が確認できるようになると各クラスターがどのような特徴を持っているか考察していくことが可能です。ぜひチャレンジしてみましょう。
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB seurat 4.3.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
scRNA-seqデータのマーカー遺伝子の特定について
SeuratにおけるFindMarkers, FindAllMarkers, FindConservedMarkers関数というマーカー遺伝子特定方法があります。それぞれ異なる目的や状況に対応するためのものです。
FindMarkers:- この関数は、特定のクラスタと他のすべてのクラスタや、2つの特定のクラスタ間のマーカー遺伝子を検出するためのものです。
ident.1とident.2という2つのパラメータを使って、比較するクラスタを指定します。- 例: クラスタ0とクラスタ1の間のマーカー遺伝子を見つけたい場合は、
FindMarkers(object, ident.1 = 0, ident.2 = 1)とします。
FindAllMarkers:- この関数は、データセット内のすべてのクラスタに対してマーカー遺伝子を一度に検出します。
FindMarkers関数を繰り返し適用するのと同じですが、効率的に一度にすべてのクラスタのマーカーを取得できます。- 例:
FindAllMarkers(object)はデータセット内のすべてのクラスタのマーカー遺伝子を返します。
FindConservedMarkers:- 複数の条件や時点、あるいは実験群間で保存されているマーカー遺伝子を探すための関数です。
- 例えば、複数のサンプルや条件で一貫して表現されるマーカー遺伝子を見つけたい場合に使用します。
grouping.varパラメータを使用して、どの条件や群を基に保存されているマーカーを探すかを指定します。- 例:
FindConservedMarkers(object, grouping.var = "condition")は、異なるconditionの値に対して保存されているマーカー遺伝子を返します。
まとめると主に以下の用途で使い分けていきます。
- 2つのクラスタ間のマーカー遺伝子を知りたい場合は
FindMarkers。 - すべてのクラスタのマーカー遺伝子を知りたい場合は
FindAllMarkers。 - 複数の条件や時点間で保存されているマーカー遺伝子を知りたい場合は
FindConservedMarkers。
今回はこれらすべての関数を実際のデータを使って解説していきます。
scRNA-seqデータの準備
それではscRNA-seqデータ統合の実装方法を紹介していきます。
データセットは下記記事で紹介したものを使います。コードの詳細はこちらの記事で紹介しているので、今回はデータセットの用意までのコードを一気に示します。
library(Seurat)
library(SeuratData)
library(patchwork)
# install dataset
InstallData("ifnb")
# load dataset
LoadData("ifnb")
# split the dataset into a list of two seurat objects (stim and CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# normalize and identify variable features for each dataset independently
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
# select features that are repeatedly variable across datasets for integration
features <- SelectIntegrationFeatures(object.list = ifnb.list)
# Find integration ancher
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features)
# this command creates an 'integrated' data assay
immune.combined <- IntegrateData(anchorset = immune.anchors)
DefaultAssay(immune.combined) <- "integrated"
FindMarkers関数
特定の細胞クラスタや群と他の細胞との間の差異発現遺伝子(DEGs)を特定するために使用されます。この関数の主な目的は、特定の細胞の状態、タイプ、または他の任意の分類に関連する遺伝子の発現の変化を明らかにすることです。
FindMarkers関数は以下2つのパターンに使用することが可能です。
- 特定の2つのクラスタ(またはクラスタのセット)間のDEGを探索
- 特定のクラスターとその他すべてのクラスタとの間のDEGを探索
それぞれ見ていきましょう
特定の2つのクラスタ(またはクラスタのセット)間のDEGを探索
こちらのケースでは2つの特定のクラスタ間の差異発現遺伝子を明確に知りたい場合に適しています。特定のクラスター間の差異を緻密に調査することが可能です。
例えば、T細胞とB細胞、あるいは健康な細胞と疾患細胞のような、2つの明確に異なる細胞集団の間の遺伝子発現の違いに特に興味がある場合、このオプションが役立ちます。
例)CD14 Mono VS CD16 Mono
以下のコードを実行して特定のクラスタペアの比較を詳細に調査することができます。
ident とはseuratパッケージでは、特定の細胞の「アイデンティティ」またはクラスタを指定するために使用されます。FindMarkers関数の場合、ident.1とident.2の引数が導入されており、これにより2つの異なるクラスタや細胞群の間の差異発現遺伝子を比較することができます。
こちらのコードはクラスター5をクラスター0と3で比較しています。
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(immune.combined, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)実行すると以下のような出力が返ってきます。
p_val avg_log2FC pct.1 pct.2 p_val_adj
CXCL10 0 -6.169698 0.555 0.951 0
CCL5 0 4.553574 0.897 0.474 0
HLA-DRA 0 -3.732949 0.459 0.986 0
FCER1G 0 -4.505242 0.213 0.869 0
IFITM3 0 -4.493810 0.577 0.926 0
それぞれの列には意味について解説していきます。
- p_val: p値は、特定の遺伝子が2つのクラスタ間で有意に差異発現しているかどうかを判断するための統計的な指標です。p値が小さいほど、遺伝子の発現に有意な差があることが示されます。
- avg_log2FC (Average Log2 Fold Change): 平均対数2倍数変化は、2つのクラスタ間の遺伝子発現の相対的な変化を示します。正の値は、
ident.1で指定されたクラスタでの発現がident.2に比べて高いことを示し、負の値はその逆を示します。 - pct.1 および pct.2 これらは、特定の遺伝子が
ident.1およびident.2のそれぞれのクラスタ内の何パーセントの細胞で発現しているかを示します。たとえば、pct.1が0.555の場合、55.5%のident.1の細胞でその遺伝子が発現していることを意味します。 - p_val_adj 補正後p値は、多重検定の補正を受けたp値を示します。これは、多くの遺伝子を同時にテストする場合の偽陽性率を考慮して調整されたものです。補正後p値が小さいほど、その遺伝子は有意に差異発現していると考えられます。
ちなみにp_valやp_val_adjが0であるのは、値がR内で設定されている浮動小数点数を超えている(限りなく0に近い)ためです。
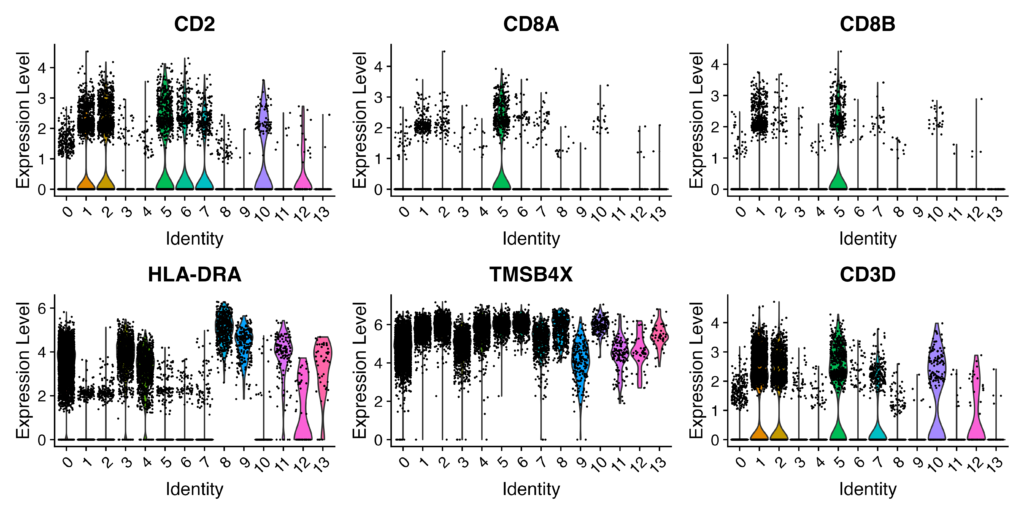
それでは実際に上位6つの遺伝子についてバイオリンプロットやドットプロットで遺伝子発現を確認してみましょう。
# トップの遺伝子を選択
top_genes <- head(row.names(cluster5.markers))
# バイオリンプロットを作成
VlnPlot(immune.combined, features = top_genes, group.by = "seurat_clusters")
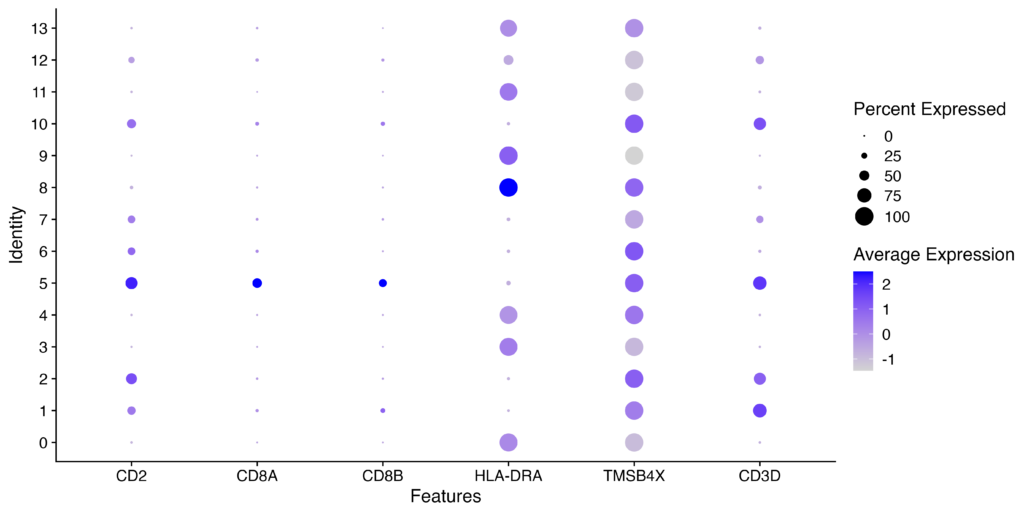
# トップの遺伝子を選択
top_genes <- head(row.names(cluster5.markers), n=5)
# ドットプロットを作成
DotPlot(immune.combined, features = top_genes, group.by = "seurat_clusters")
特定のクラスターとその他すべてのクラスタとの間のDEGを探索
例)CD14 Mono VS CD14 Mono以外のすべてのセル(CD16 Mono, CD4 Memory T, CD4 Naive T…などすべてのセル)
下記のコードクラスタ2とそれ以外のすべてのセルとの間での差異発現遺伝子を識別します。
cluster0.markers <- FindMarkers(immune.combined, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
head(cluster0.markers, n = 5)以下のような出力が返ってきます。
myAUC avg_diff power avg_log2FC pct.1 pct.2
FTL 0.975 2.294724 0.950 3.310586 1.000 0.877
CD63 0.954 1.834068 0.908 2.646001 0.994 0.324
ANXA5 0.941 1.759063 0.882 2.537791 0.987 0.322
C15orf48 0.936 1.999672 0.872 2.884916 0.986 0.193
LGALS3 0.923 2.018229 0.846 2.911689 0.953 0.239
実際に”FTL”, “CD63″についてバイオリンプロットで発現を確認してみましょう。
VlnPlot(immune.combined, features = c("FTL", "CD63"), pt.size = 0.1, group.by = "seurat_clusters")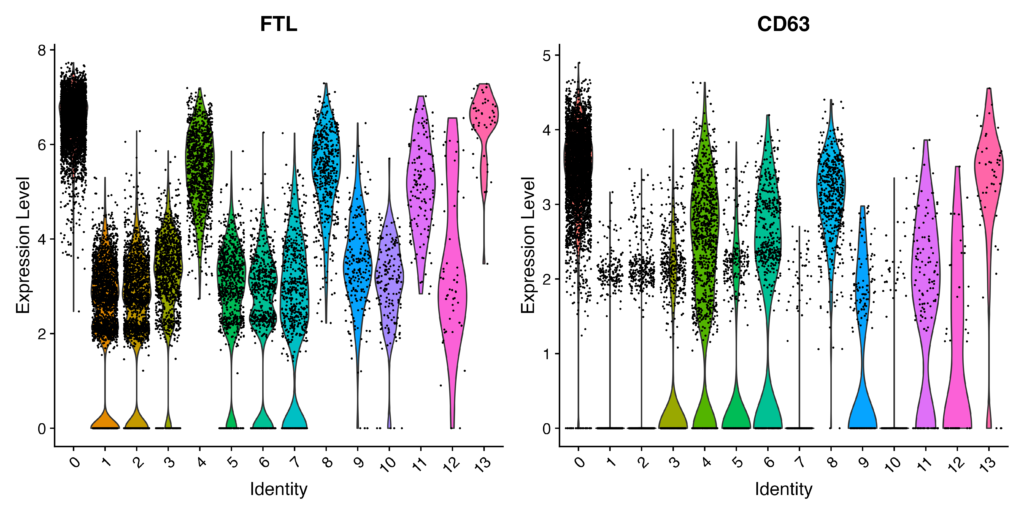
クラスター0に置いて、FTL, CD63の発現が他よりも高く出ていることや多くの細胞(黒の点)で高発現が確認できます。FTL, CD63はクラスター0のマーカー遺伝子として有意に機能しそうです。
FindAllMarkers関数
先程のFindMarkers関数では特定のクラスタのペア間の詳細な比較を行いたい場合や特定のクラスターとその他すべてのクラスタとの比較の場合に使用可能でした。ある特定のクラスターを指定する分詳細な解析は可能ですが、クラスター分関数を適用するのは大きな手間になります。
FindAllMarkersを実行すると、全体的な差異を迅速に把握することができます。つまり、Seuratオブジェクト内のすべてのクラスタに対して、そのクラスタのセルと他のすべてのセルとの間での差異発現遺伝子(DEG)を同時に探索します。
# Identify markers for every cluster compared to all remaining cells, report only the positive ones
cluster.markers <- FindAllMarkers(immune.combined, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
cluster.markers %>%
group_by(cluster) %>%
slice_max(n = 2, order_by = avg_log2FC) %>%
print(n=28)上記を実行すると以下のような出力が出てきます。
# A tibble: 28 × 7
# Groups: cluster [14]
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 0 5.39 0.96 0.537 0 0 CCL8
2 0 5.36 0.987 0.512 0 0 CCL2
3 0 2.34 0.929 0.659 0 1 SELL
4 0 1.93 0.98 0.685 0 1 GIMAP7
5 0 1.82 0.785 0.31 0 2 ALOX5AP
6 0 1.70 0.743 0.379 0 2 TRAT1
7 4.30e- 8 3.61 0.285 0.085 8.59e- 5 3 IGLL5
8 0 3.41 0.734 0.128 0 3 CD79A
9 0 4.31 0.774 0.187 0 4 VMO1
10 0 3.94 0.992 0.314 0 4 FCGR3A
11 0 2.97 0.897 0.418 0 5 CCL5
12 1.16e-241 2.86 0.77 0.432 2.32e-238 5 LAG3
13 0 6.08 0.975 0.35 0 6 GNLY
14 0 4.56 0.944 0.344 0 6 GZMB
15 2.03e- 95 2.60 0.773 0.563 4.07e- 92 7 GADD45B
16 1.20e- 96 2.46 0.797 0.609 2.39e- 93 7 CD69
17 8.79e-181 3.11 0.998 0.67 1.76e-177 8 TXN
18 1.49e-277 3.10 0.998 0.541 2.99e-274 8 HLA-DPB1
19 1.73e-133 4.46 0.941 0.451 3.46e-130 9 MIR155HG
20 5.76e- 95 3.71 0.882 0.313 1.15e- 91 9 MYC
21 1.38e-105 5.15 0.92 0.282 2.75e-102 10 PPBP
22 1.18e- 97 3.78 0.823 0.253 2.36e- 94 10 PF4
23 2.57e-163 4.54 0.917 0.149 5.15e-160 11 TSPAN13
24 5.62e- 47 3.73 0.977 0.678 1.12e- 43 11 TXN
25 6.05e- 39 10.4 1 0.386 1.21e- 35 12 HBB
26 2.98e- 45 9.63 1 0.276 5.96e- 42 12 HBA2
27 3.93e- 32 3.74 0.979 0.287 7.87e- 29 13 PPBP
28 5.05e- 25 2.78 0.851 0.233 1.01e- 21 13 GNG11
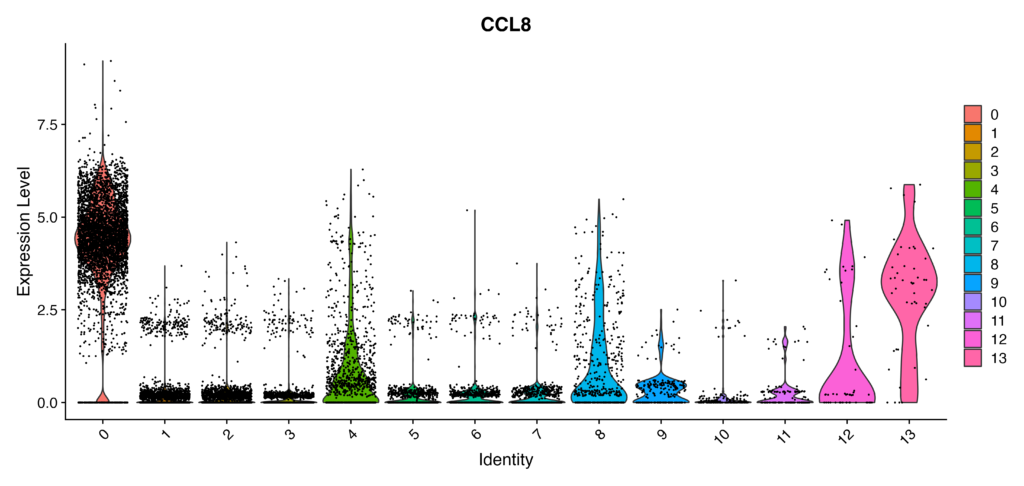
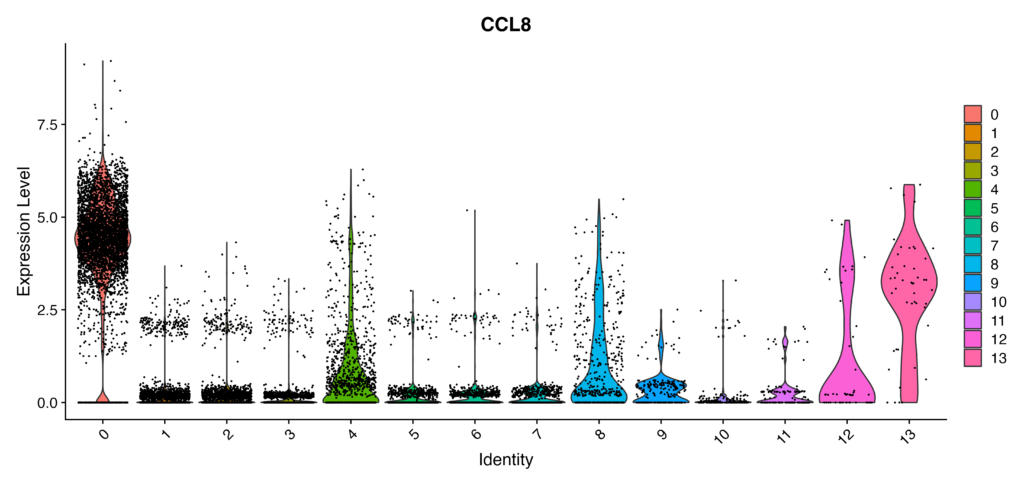
では実際にマーカー遺伝子発現を可視化してみましょう。まずはCCL8をバイオリンプロットでみてみます。
plots <- VlnPlot(immune.combined, features = c("CCL8"), group.by = "seurat_clusters", pt.size = 0.1, combine = FALSE)こちらのような出力が出れば成功です。クラスター0のExpression Levelが他のクラスターに比べて高いのが確認できます。
FindAllMarkerのような多くのマーカー遺伝子が検出される場合はDotplotの方がすべてのマーカー遺伝子を一覧できるので向いています。
marker_genes <- cluster.markers %>%
group_by(cluster) %>%
slice_max(order_by = avg_log2FC, n = 2) %>%
pull(gene)
unique_marker_genes <- unique(marker_genes)
DotPlot(immune.combined, features = unique_marker_genes, group.by = "seurat_clusters") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1))それぞれのクラスターでマーカー遺伝子として発現が高いのが確認できます。
FindConservedMarkers関数
コントロール群と刺激群などを統合したデータセット間において保存されているマーカー遺伝子はFindConservedMarkers()関数で同定していきます。これにより、刺激条件が変わっても一貫して発現しているマーカー遺伝子を、特定の細胞クラスターに対して特定できます。この情報は、細胞タイプの識別や機能解析に役立ちます。FindMarkers 関数は、単一の条件や単一の実験データセット内の2つのクラスタ間の差異発現遺伝子を見つけるために使用されますが、FindConservedMarkers関数は、複数の条件や複数の実験データセットを考慮して、一貫して差異発現する遺伝子を見つけるために使用されます。例えば、健康な個体と疾患を持つ個体の2つの異なるデータセットで、特定のクラスタのセルで一貫して上昇または低下している遺伝子を見つけたい場合に使用します。
以下のコードを実行していきます。
# For performing differential expression after integration, we switch back to the original
# data
DefaultAssay(immune.combined) <- "RNA"
cluster6.markers <- FindConservedMarkers(immune.combined, ident.1 = 6, grouping.var = "stim")
head(cluster6.markers)DefaultAssay(immune.combined) <- "RNA": 統合データセットimmune.combinedのデフォルトのアッセイ(解析対象とするデータレイヤー)を"RNA"に戻します。これは、元のRNA-seqデータを基に差異発現解析を行うためです。FindConservedMarkers: この関数は、特定の細胞アイデンティティ(ここではident.1 = 6で指定されている)で、特定のグループ変数(ここでは"stim"で刺激の有無が格納されている)によって一貫して発現しているマーカー遺伝子を探します。head(nk.markers): 見つかったマーカー遺伝子のリストの先頭部分を表示します。
上記のコード実行すると以下のような出力が現れると思います。
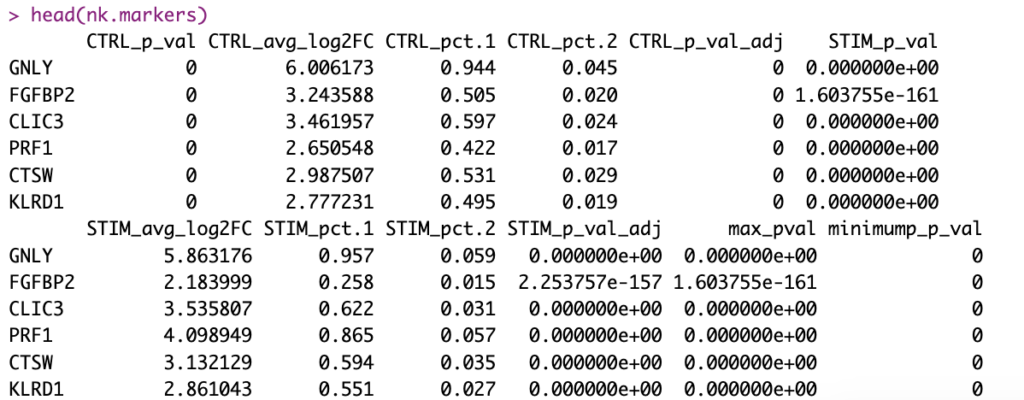
FindConservedMarkers()関数の出力は、指定したクラスターに対する遺伝子IDでリスト化された潜在的なマーカーの順位付けされたリストと、関連する統計情報を含む行列です。注意点として、各グループ(この場合、CtrlとStim)に対して同じ統計セットが計算され、最後の2列は2つのグループ全体での統合されたp値に対応します。以下は、いくつかの列の説明です。(conditionは、CTRLとSTIMを指します)
- gene: 遺伝子シンボル
- 〇〇_p_val: 条件に対して複数のテスト修正が調整されていないp値
- 〇〇_avg_logFC: 条件に対する平均ログフォールド変化。正の値は、その遺伝子がクラスタ内で高く発現していることを示す。
- 〇〇_pct.1: 条件において、クラスタ内で遺伝子が検出される細胞の割合
- 〇〇_pct.2: 条件において、他のクラスターでの平均として遺伝子が検出される細胞の割合
- 〇〇_p_val_adj: データセット内の全遺伝子を使用してボンフェロニ補正に基づく調整されたp値で、有意性を決定するために使用される
- max_pval: 各グループ/条件で計算されたp値の最大値
- minimump_p_val: 統合されたp値
出力を見るときは、 pct.1とpct.2の間の発現の差が大きく、fold changesが大きいマーカーを探すことをお勧めします。例えば、pct.1= 0.90、pct.2= 0.80の場合、それほど有効なマーカーではないかもしれませんが、代わりにpct.2=0.1であれば、差が大きく説得力を持ちます。pct.1=0.3など低ければ、それほど興味深いマーカーでない可能性が高いです。
Granulysin (GNLY)は、NK細胞で特異的な発現が報告されている細胞傷害性タンパク質で知られています。GNLYコントロール群と刺激群でConserveされているマーカーなのでクラスター6はNK細胞として割り当てられるでしょう。実際に、featureプロットでGNLYと他の免疫細胞のマーカーも比べてみましょう。
FeaturePlot(immune.combined, features = c("CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A","CCL2", "PPBP"), min.cutoff = "q9")
Featureplotで実際のマーカー遺伝子発現がクラスター特異的に確認できたら、RenameIdents関数でそれぞれのクラスターに対して、細胞種を割り当てます。
immune.combined <- RenameIdents(immune.combined, `0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T", `3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "NK", `7` = "T activated", `8` = "DC", `9` = "B Activated",`10` = "Mk", `11` = "pDC", `12` = "Eryth", `13` = "Mono/Mk Doublets")
DimPlot(immune.combined, label = TRUE)
補足
FindConservedMarkers関数を使う前にバッチエフェクトを補正しておくことが大事です。複数の条件または実験グループを比較する場合、バッチエフェクトが補正されていないと、バッチ由来の変動が実際の生物学的な違いと誤解されるリスクがあります。SeuratパッケージはIntegrateData関数など、複数のデータセットを統合し、バッチエフェクトを補正するためのツールを提供していますので予め適応しておきましょう。
最後に
いかがだったでしょうか。マーカー遺伝子の発現が確認できるようになると各クラスターを特徴づけて考察していくことができます。ぜひ挑戦してみてください。
参考
Single-cell RNA-seq marker identification
Finding differentially expressed features (cluster biomarkers)
Introduction to scRNA-seq integration

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

