
ikraやiDEPが使えるようにはなってきたが、もっと柔軟に解析したい!という方もいらっしゃのではないでしょうか。Rを使えば、もっと柔軟に、さらにきれいなバイオインフォマティクス向けの図の作成が可能です。バイオインフォマティクスで最初に可視化させる方法として主成分分析解析を紹介したいと思います。ぜひチャレンジしてみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
主成分分析(PCA)とは?
主成分分析とは、多次元データを低次元に次元圧縮する手法の一つです。主成分分析は、データの性質を最もよく表す要素を抽出することができるため、データの可視化やクラスタリングなどの機械学習手法を用いた分析にも活用されます。
RNA-seq解析では、主成分分析を使用することで、大量の発現データをより簡単かつ高速に解析することができます。主成分分析は、原発現データの次元圧縮を行い、主な発現変動のパターンを抽出します。これにより、大量の発現データを分かりやすく視覚化し、その結果をもとに遺伝子の発現変動や発現パターンを解析することができます。
Rを実行する準備
今回はRstudioを使ってRを実行していきます。
各OS環境を元にこちらのサイトよりインストーラーをダウンロードしてください。
https://cran.r-project.org/
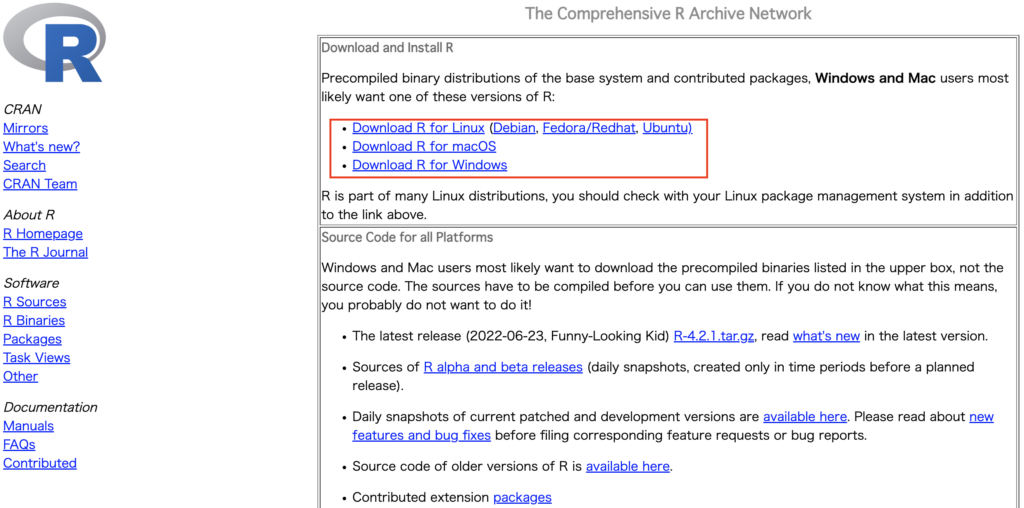
インストールするバージョンは最新版を選んでください。一番トップにあるものが最新版です。

インストール画面では「続ける」を押して完了させてください。
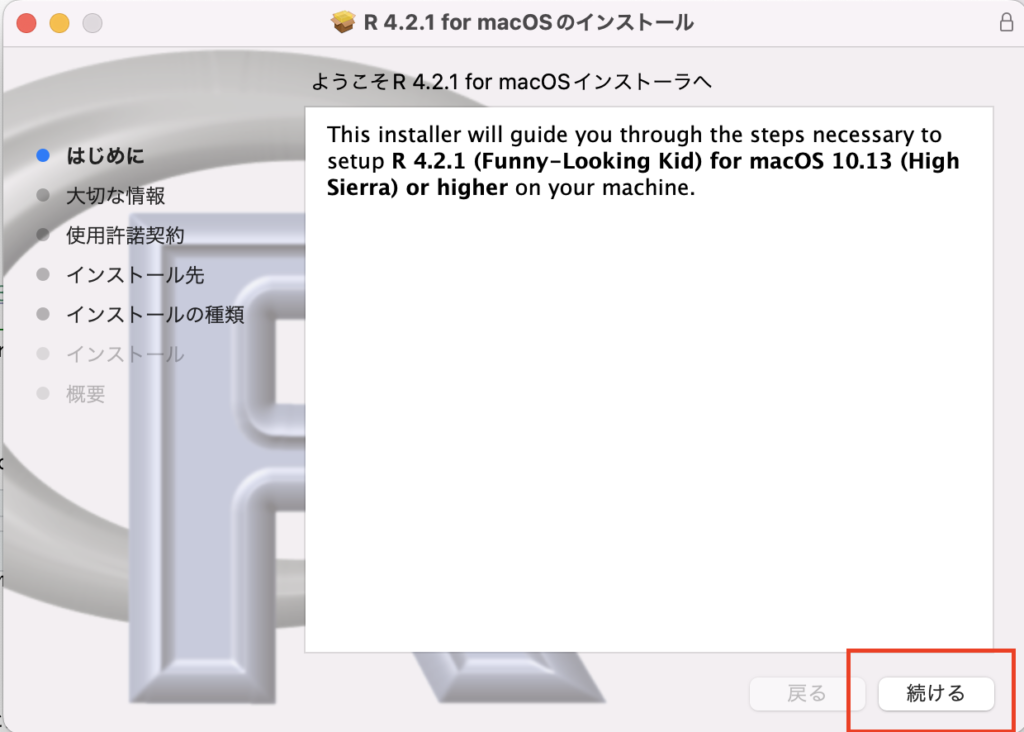
Rstudioでは左上のエリアにコードを書いていきます。
Runを押すと行実行ができます。
Sourceを押すとスクリプト全体が実行できますので覚えておいてください。
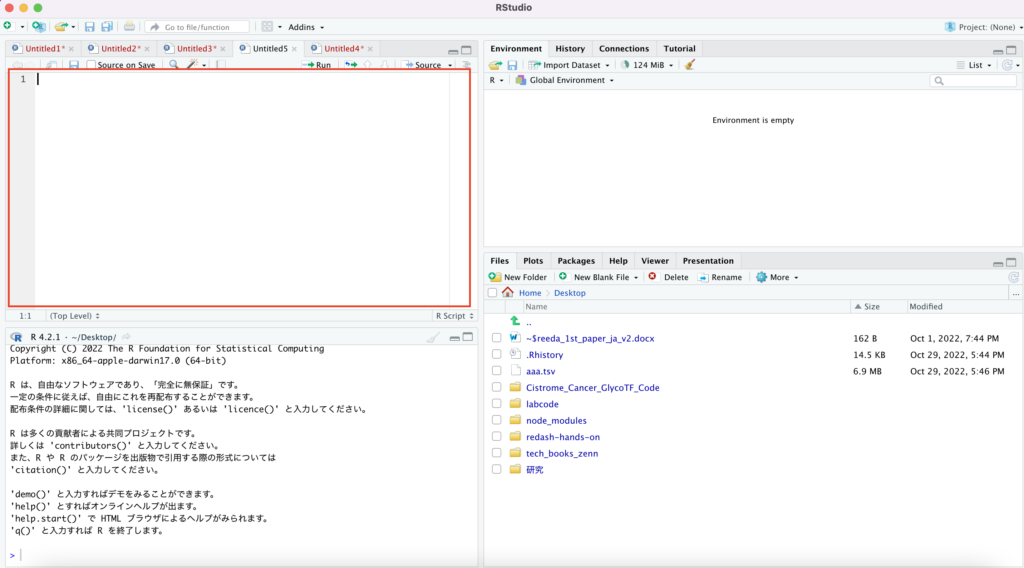
次にDesktopを作業ディレクトリにするためにSet As Working Directory作業を行います。Rstudioの右下画面でデスクトップまで進み、
- Moreをクリック
- Set As Working Directory
を順に選びます。
この作業を間違うと後のコードが一切通らなくなるのでお気をつけください。
以下のコマンドを叩いて最後にDesktopで終わるようなパスが出てきたらきちんと設定がなされています。
> getwd()
[1] "/Users/[ユーザー名]/Desktop"Rを用いてRNA−seqデータを主成分分析する方法
Rを用いてRNA−seqデータを主成分分析するには以下のようなコードを書きます。まずはこちらのコードを保存しましょう。
install.packages("ggplot2")
library(ggplot2)
install.packages("devtools")
library(devtools)
install_github('sinhrks/ggfortify')
library(ggfortify)
library(tidyverse)
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 2)
mydata = read_tsv('sample.tsv')
t_mydata=as.data.frame(t(mydata))
gene_names=t_mydata[1,]
colnames(t_mydata) <- gene_names
delete_data <- c()
for(x in t_mydata) {
data5 <- sapply(x[2:length(x)], as.numeric)
data5_var <- var(data5)
if (data5_var == 0) {
delete_data <- c(delete_data, x[1])
}
}
for(x in delete_data){
t_mydata <- select(t_mydata, -x)
}
t_mydata_as_num <- sapply(t_mydata, as.numeric)
data2 <- na.omit(t_mydata_as_num)
pca_res <- prcomp(data2, scale. = TRUE)
autoplot(pca_res)コードはFile→Save Asと進み、名前をつけたらWhereを「Desktop」にして保存しましょう。
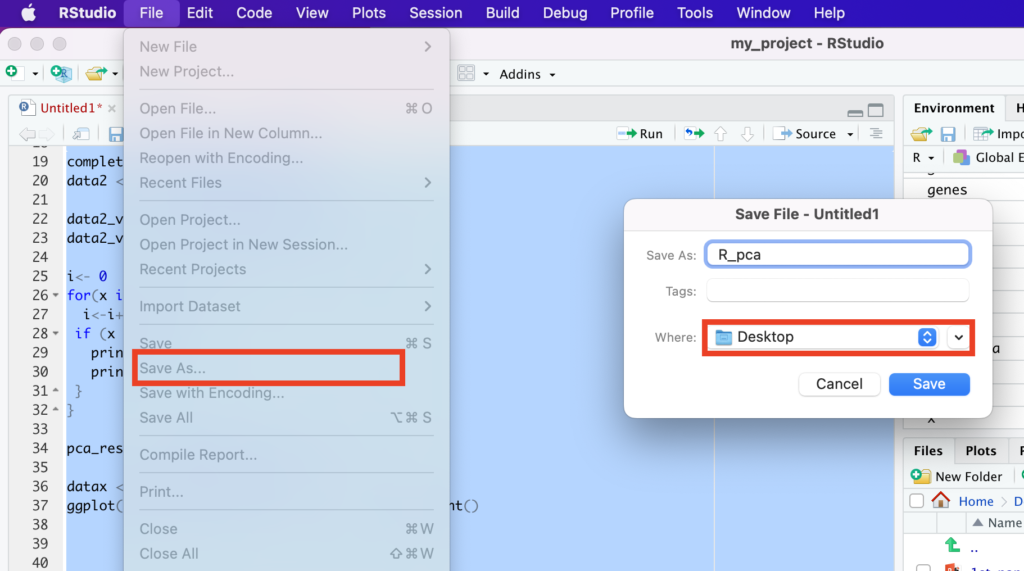
実際にコードを実行してみよう
コードを実行する前に解析データを用意します。実際に解析に使うファイルはこのようなマトリクスファイルを作成するイメージです。
| SRR11111111 | SRR11111112 | SRR11111113 | …… | |
|---|---|---|---|---|
| A1BG | 1.204012121 | 1.305946294 | 1.058155716 | |
| A1CF | 0.8814023041 | 1.010914863 | 1.174708571 | |
| A2M | 1.868867371 | 1 | 0.9556968534 | |
| A2ML1 | 0.6709431377 | 1.126605147 | 1 | |
| A3GALT2 | 1 | 1 | 1 | |
| A4GALT | 0.5010093702 | 1.364249746 | 1.664916037 | |
| A4GNT | 1.881158763 | 1 | 1 | |
| AAAS | 0.7203903135 | 0.8684943431 | 1.474680837 | |
| AACS | 1.06245034 | 1.256530797 | 1.544221892 | |
| (以下略) |
今回はサンプルファイルを用意しましたのでこちらをデスクトップにダウンロードしてお使いください。
実際にサンプルファイルを読み込んでSourceを押してコードを実行してみると以下のような分析結果になると思います。
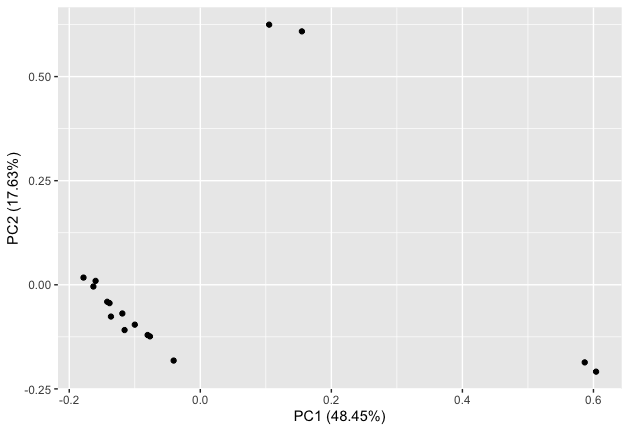
ここからサンプルごとに色を塗り分けてみようと思います。
以下のコマンドをRstudio左下のコマンドラインでたたき、sample_2.tsv ファイルを作ってください。
write_tsv(as.data.frame(data2), "sample_2.tsv")1列目にSampleという列を作ってください。テーブルを貼っておきますのでコピーしてご活用ください。
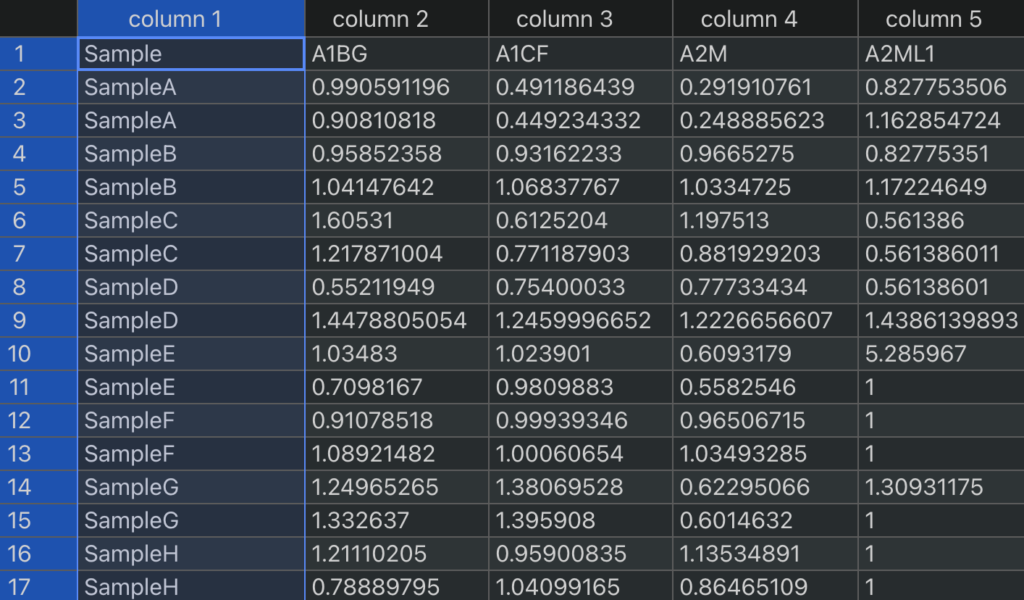
| Sample |
| SampleA |
| SampleA |
| SampleB |
| SampleB |
| SampleC |
| SampleC |
| SampleD |
| SampleD |
| SampleE |
| SampleE |
| SampleF |
| SampleF |
| SampleG |
| SampleG |
| SampleH |
| SampleH |
準備ができたら以下のコードを実行してみましょう。デスクトップにsample_2.tsvが置かれていないとエラーになるので気をつけてください。
mydata = read_tsv('sample_2.tsv')
pca_res <- prcomp(mydata[2:ncol(mydata)], scale. = TRUE)
autoplot(pca_res, data=mydata, colour="Sample")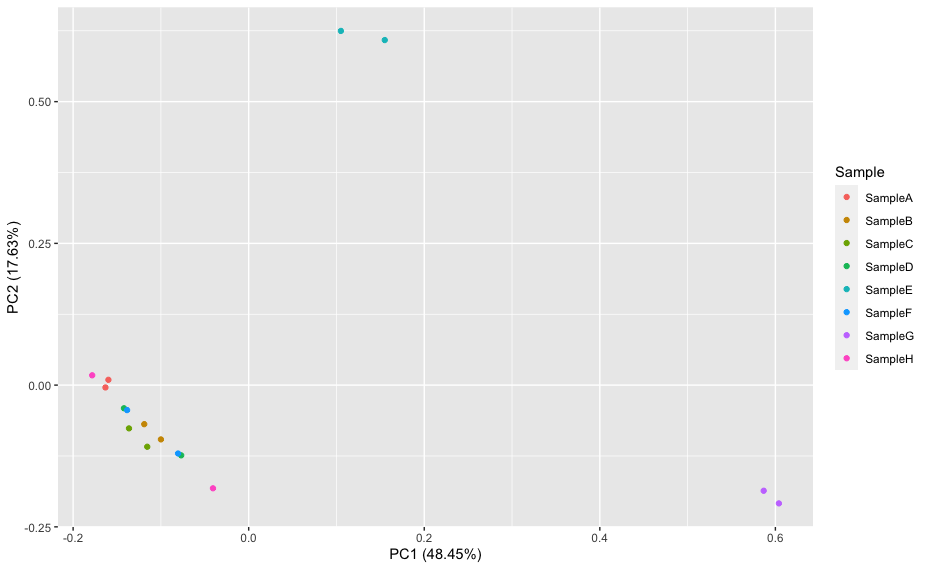
サンプルごとに分けることができました。
コードの解説
以下のコードで、PCAに必要なパッケージをインストールしています。
install.packages("ggplot2")
library(ggplot2)
install.packages("devtools")
library(devtools)
install_github('sinhrks/ggfortify')
library(ggfortify)
library(tidyverse)以下は、Rstudioに読み込むデータ容量を上げています。
今回のようにサイズが小さいファイルなら問題ありませんが、大きいファイルは読み込むことができなくなるので以下のコマンドを叩かないとエラーになります。
Sys.setenv("VROOM_CONNECTION_SIZE" = 131072 * 2)read_tsv()関数を使用して’sample.tsv’という名前のtsvファイルからデータを読み込み、変数’mydata’に格納します。次に、as.data.frame()関数とt()関数を使用して、’t_mydata’というデータフレームに’mydata’を転置します。
mydata = read_tsv('sample.tsv')
t_mydata=as.data.frame(t(mydata))gene_namesにはサンプル名を代入します。次に、colnames()関数を使用して、’t_mydata’の列名をgene_namesに設定します。
gene_names=t_mydata[1,]
colnames(t_mydata) <- gene_namesこちらのコードは詳しい説明は割愛しますが、端的に説明すると、主成分分析では単位分散(今回の場合、発現量がすべて1の遺伝子)の成分を除く必要があり、こちらのコードで除いています。
delete_data <- c()
for(x in t_mydata) {
data5 <- sapply(x[2:length(x)], as.numeric)
data5_var <- var(data5)
if (data5_var == 0) {
delete_data <- c(delete_data, x[1])
}
}
for(x in delete_data){
t_mydata <- select(t_mydata, -x)
}こちらではデータから欠損値を削除しています。削除されたデータはdata2に格納します。
t_mydata_as_num <- sapply(t_mydata, as.numeric)
data2 <- na.omit(t_mydata_as_num)最後にprcomp関数で主成分分析の実行です。引数にscale. = TRUEを指定し、正規化を行ってから主成分分析をすることができます。
最後にautoplot関数によって可視化をしています。
pca_res <- prcomp(data2, scale. = TRUE)
autoplot(pca_res)autoplotでcolourを指定し、グループ化したい列を指定すると、そちらでグループ化できます。
グループ化したい列は文字列になっているので、mydata[2:ncol(mydata)]で文字列を避けてPCAを行っています。そのままやるとエラーになるので気をつけてください。
pca_res <- prcomp(mydata[2:ncol(mydata)], scale. = TRUE)
autoplot(pca_res, data=mydata, colour="Sample")おわり
いかがだったでしょうか。PCAのような次元圧縮法はRNA−seq解析のような多次元の要素を含むサンプルをあつかう上で有用な解析です。Rを覚えはじめとしてPCAに挑戦してみるのはいかがでしょうか。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます


1列目にSampleという列を作ってください。テーブルを貼っておきますのでコピーしてご活用ください。
ここができません、よろしければ詳細説明をお願い致します。
稲村 和紀様
ご質問ありがとうございます。
write_tsvコマンドで “sample_2.tsv”を作成された後に、どのツールでもよいのですが、例えばVSCodeやExcelなどで開いていただき、一列目に、Sample列を新しく作成していただくという内容になります。
よろしくお願い致します。