
single cell RNA-seq解析はある程度できるようになったけど、複数のデータセットを扱った解析はまだできないという方はいるのではないでしょうか。
この記事ではsingle cell RNA-seq解析の複数のデータセットを統合する方法を紹介します。
これにより、複数の実験から得られたsingle cell RNA-seq解析データを扱うことができるようになります。ぜひ挑戦してみましょう。
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB seurat 4.3.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
scRNA-seqデータセットを結合とは?
scRNA-seqデータセットを結合とは、2つ以上の独立したscRNA-seqデータセットを1つの統合データセットにまとめるプロセスを指します。わざわざそれぞれ取得したデータをわざわざ結合させるのはなぜ..?と思うかもしれませんが大きく分けて2つの理由があります。
一つ目は、異なる時点や条件のサンプルをまとめるためです。研究者は時間経過や異なる実験条件下でのサンプルからの細胞の変動を捉えるために、複数のサンプルをシーケンスすることがあります。これらのサンプルを結合することで、全体としての動的な変化や条件間の違いを捉えることができます。
2つ目は、技術的な理由で、一度にシーケンスできる細胞の数には上限があり、大量の細胞を研究するためには複数のデータセットに分けてシーケンスする必要があります。
merge関数について
merge関数は、特にSeuratパッケージのコンテキストで言うと、複数のSeuratオブジェクトを1つのオブジェクトに結合するための関数です。このmerge関数を使用することで、複数のデータセットを1つのデータセットとして扱うことが可能になります。
バッチエフェクト補正まで含めると統合であり、今回はseuratobjectを結合させているだけなのでバッチエフェクト問題が解消されていないことは注意が必要です。mergeは、バッチエフェクトが少ない、またはその影響が許容範囲内であると考えられる場合のシンプルなデータ結合に用いると良いでしょう。
データセットの準備
それでは実際にデータセットの統合を試してみたいと思います。
今回はこちらのハンズオンをベースに解説していきます。
まずは以下のデータセットにアクセスしてください。
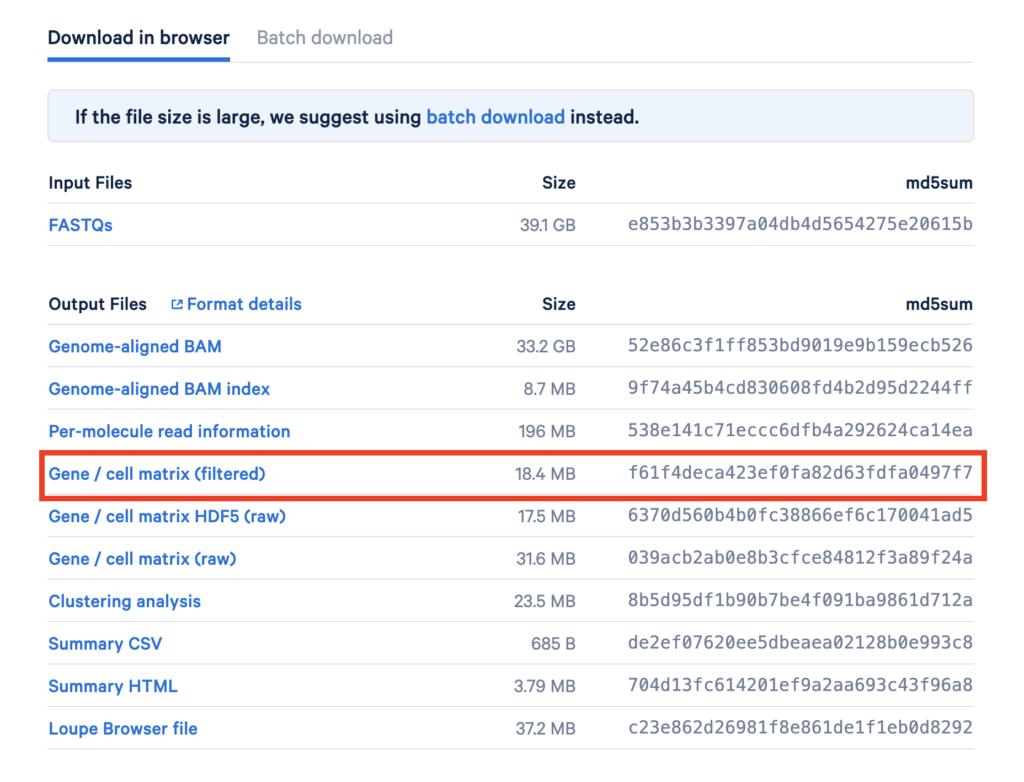
下にスクロールすると画像赤枠のGene / cell matrix(filterd)があると思いますのでそれぞれのデータセット分ダウンロードしてください。
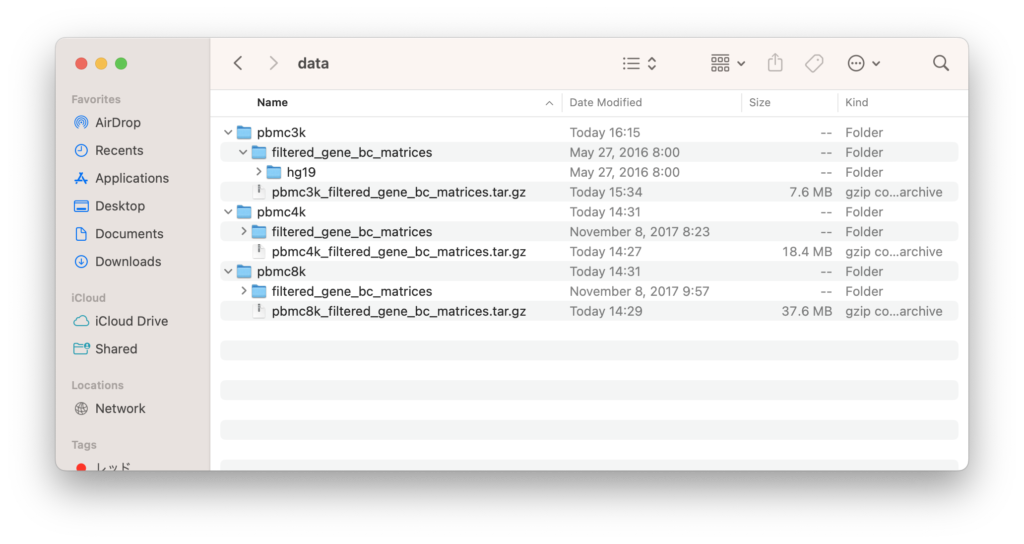
デスクトップでdataフォルダを作成し、以下の画像の様なディレクトリ構成にしてください。各フォルダにあるfilterd_gene_bc_matricesは、gzipファイルを解凍すると作成されます。ディレクトリのパスとしては以下になります。
# pbmc4k
"./data/pbmc4k/filtered_gene_bc_matrices/GRCh38/"
# pbmc8k
"./data/pbmc8k/filtered_gene_bc_matrices/GRCh38/"
# pbmc3k
"./data/pbmc3k/filtered_gene_bc_matrices/hg19/"
ここまでできれば準備完了です。
2つのscRNA-seqデータセットを結合する
それでは実際にscRNA-seqデータ同士を結合させてみましょう。結合前と結合後が分かるように都度出力を確認しながら進んでいきます。
まずは、pbmc4k を確認していきます。
pbmc4k.data <- Read10X(data.dir = "./data/pbmc4k/filtered_gene_bc_matrices/GRCh38/")
pbmc4k <- CreateSeuratObject(counts = pbmc4k.data, project = "PBMC4K")
pbmc4k
pbmc4k の出力は以下です。4340 samples 含まれるようです。
> pbmc4k
An object of class Seurat
33694 features across 4340 samples within 1 assay
Active assay: RNA (33694 features, 0 variable features)
次には、pbmc8k を確認していきます。
pbmc8k.data <- Read10X(data.dir = "./data/pbmc8k/filtered_gene_bc_matrices/GRCh38/")
pbmc8k <- CreateSeuratObject(counts = pbmc8k.data, project = "PBMC8K")
pbmc8kpbmc8k の出力は以下です。8381 samples 含まれるようです。
## > pbmc8k
## An object of class Seurat
## 33694 features across 8381 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
それでは、pbmc4kとpbmc8kを結合していきましょう。
以下のようなコードになります。
pbmc.combined <- merge(pbmc4k, y = pbmc8k, add.cell.ids = c("4K", "8K"), project = "PBMC12K")
pbmc.combined
このコードは、2つのscRNA-seqデータセット(pbmc4k と pbmc8k)をSeuratパッケージの merge 関数を使用して結合し、新しいデータセット pbmc.combined を作成しています。データセットの結合を行っています。初めて見る引数について解説していきます。
- y = pbmc8k: 結合する2つ目のSeuratオブジェクト。
y引数で指定されています。 - add.cell.ids = c(“4K”, “8K”): 結合するデータセットの細胞に接頭辞を追加して、元々のデータセットを識別できるようにするための引数です。ここでは、
pbmc4kからの細胞には “4K” という接頭辞が、pbmc8kからの細胞には “8K” という接頭辞が追加されます。これにより、結合後のデータセット内で元のデータセットに属していた細胞を区別することができます。
このコードを実行すると、pbmc.combined という新しいSeuratオブジェクトが作成され、このオブジェクトには pbmc4k と pbmc8k の両方のデータセットからの細胞と遺伝子発現データが含まれます。本当に結合されたかpbmc.combined の出力を確認していきましょう。
pbmc.combined の出力は以下です。12721 samples 含まれることが確認できます。
4340 (pbmc4k) + 8381 (pbmc8k) = 12721 samples (pbmc.combined) になっていることが確認できます。実際に結合されたことが確認できますね。
## > pbmc.combined
## An object of class Seurat
## 33694 features across 12721 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
結合後にセルの識別子が正しく追加されていることを確認していきます。
head(colnames(pbmc.combined))
tail(colnames(pbmc.combined))出力をそれぞれ見ると、セルの名前に"4K_", "8K_"というプレフィックスが追加されていることが分かります。これにより、この特定のセルがどちらのデータセットからのものであることが分かります。
> head(colnames(pbmc.combined))
[1] "4K_AAACCTGAGAAGGCCT-1" "4K_AAACCTGAGACAGACC-1" "4K_AAACCTGAGATAGTCA-1"
[4] "4K_AAACCTGAGCGCCTCA-1" "4K_AAACCTGAGGCATGGT-1" "4K_AAACCTGCAAGGTTCT-1"
> tail(colnames(pbmc.combined))
[1] "8K_TTTGTCAGTTACCGAT-1" "8K_TTTGTCATCATGTCCC-1" "8K_TTTGTCATCCGATATG-1"
[4] "8K_TTTGTCATCGTCTGAA-1" "8K_TTTGTCATCTCGAGTA-1" "8K_TTTGTCATCTGCTTGC-1"
pbmc4k と pbmc8kの数を集計してみましょう。集計するときはtable関数を使います。
table(pbmc.combined$orig.ident)4340 (pbmc4k) + 8381 (pbmc8k)とそれぞれのカウント数が返ってきました。先程確認した値と一致しますね。
> table(pbmc.combined$orig.ident)
PBMC4K PBMC8K
4340 8381
2つ以上のscRNA-seqデータセットを結合する
先程のセクションで2つのscRNA-seqデータセットが結合できるようになりました。それでは、2つ以上のデータセットの統合はどのようにやるのでしょうか?基本的には2つでも3つでもmerge関数を結合させるのは変わりません。実際にやってみましょう。
まずは第三のデータセット pbmc3k を読み込んでいきます。
pbmc3k.data <- Read10X(data.dir = "./data/pbmc3k/filtered_gene_bc_matrices/hg19/")
pbmc3k <- CreateSeuratObject(counts = pbmc3k.data, project = "PBMC3K")
pbmc3kpbmc4k の出力は以下です。2700 samples 含まれるようです。
## > pbmc3k
## An object of class Seurat
## 32738 features across 2700 samples within 1 assay
## Active assay: RNA (32738 features, 0 variable features)
それではmerge関数で3つのデータセットを結合させていきます。先程と異なるのはyに2つのデータセット(pbmc4k, pbmc8k)とadd.cell.idsに3つの識別子(”3K”, “4K”, “8K”)を記載するようにしただけです。
pbmc.big <- merge(pbmc3k, y = c(pbmc4k, pbmc8k), add.cell.ids = c("3K", "4K", "8K"), project = "PBMC15K")
pbmc.bigpbmc.big の出力は以下です。15421 samples 含まれることが確認できます。
4340 (pbmc4k) + 8381 (pbmc8k) + 2700 (pbmc3k) = 15421 samples (pbmc.combined) になっていることが確認できます。実際に3つのデータセットが結合されたことが確認できますね。
## > pbmc.big
## An object of class Seurat
## 36116 features across 15421 samples within 1 assay
## Active assay: RNA (36116 features, 0 variable features)
結合後にセルの識別子が正しく追加されていることを確認していきます。こちらのコードで確認できます。
unique(sapply(X = strsplit(colnames(pbmc.big), split = "_"), FUN = "[", 1))以下のように”3K” “4K” “8K”がそれぞれ追加されています。
## > unique(sapply(X = strsplit(colnames(pbmc.big), split = "_"), FUN = "[", 1))## [1] "3K" "4K" "8K"
pbmc4k と pbmc8k と pbmc3k 数を集計してみましょう。
table(pbmc.big$orig.ident)それぞれ確認していた値が集計されて出力されました。
## > table(pbmc.big$orig.ident)
## PBMC3K PBMC4K PBMC8K
## 2700 4340 8381
正規化データに基づいて結合させる場合
merge関数は正規化されたデータセット同士を結合させることが可能です。統合前に正規化を行うことは、データの一貫性を保持し、後の解析ステップでの問題や誤解を避けるための一般的なベストプラクティスとして知られています。
それでは、2つのscRNA-seqデータセットを正規化し、正規化されたデータを基に統合する操作していきます。pbmc4kとpbmc8kをNormalizeData関数によって正規化した後にそれぞれを結合させていきます。
pbmc4k <- NormalizeData(pbmc4k)
pbmc8k <- NormalizeData(pbmc8k)
pbmc.normalized <- merge(pbmc4k, y = pbmc8k, add.cell.ids = c("4K", "8K"), project = "PBMC12K",
merge.data = TRUE)GetAssayData関数は特定のアッセイの特定のデータを閲覧することができる関数でseuratオブジェクトの中のデータを見ることができます。
GetAssayData(pbmc.normalized)[1:10, 1:15]以下のような出力になります。これだけでは分かり辛いので正規化前(pbmc.combined)も同様に出力してみましょう。
## > GetAssayData(pbmc.normalized)[1:10, 1:15]
## 10 x 15 sparse Matrix of class "dgCMatrix"
## [[ suppressing 15 column names ‘4K_AAACCTGAGAAGGCCT-1’, ‘4K_AAACCTGAGACAGAC## C-1’, ‘4K_AAACCTGAGATAGTCA-1’ ... ]]
## RP11-34P13.3 . . . . . . . . . . . . . . .
## FAM138A . . . . . . . . . . . . . . .
## OR4F5 . . . . . . . . . . . . . . .
## RP11-34P13.7 . . . . . . . . . . . . . . .
## RP11-34P13.8 . . . . . . . . . . . . . . .
## RP11-34P13.14 . . . . . . . . . . . . . . .
## RP11-34P13.9 . . . . . . . . . . . . . . .
## FO538757.3 . . . . . . . . . . . . . . .
## FO538757.2 . . . . . . . . . 0.7721503 . . . . .
## AP006222.2 . . . . . . . . . . . 1.087928 . . .
GetAssayData(pbmc.combined)[1:10, 1:15]正規化前(pbmc.combined)の出力です。
生のカウントデータは通常、非負の整数のみを含みます。一方、正規化されたデータは、浮動小数点の数値に変換されることがよくあります。また、正規化されたデータは、0より大きい浮動小数点の値を持つことが一般的です。先程の正規化後(pbmc.normalized)と比較するとそれらが確認できるでしょう。
## > GetAssayData(pbmc.combined)[1:10, 1:15]
## 10 x 15 sparse Matrix of class "dgCMatrix"
## [[ suppressing 15 column names ‘4K_AAACCTGAGAAGGCCT-1’, ‘4K_AAACCTGAGACAGAC## C-1’, ‘4K_AAACCTGAGATAGTCA-1’ ... ]]
## RP11-34P13.3 . . . . . . . . . . . . . . .
## FAM138A . . . . . . . . . . . . . . .
## OR4F5 . . . . . . . . . . . . . . .
## RP11-34P13.7 . . . . . . . . . . . . . . .
## RP11-34P13.8 . . . . . . . . . . . . . . .
## RP11-34P13.14 . . . . . . . . . . . . . . .
## RP11-34P13.9 . . . . . . . . . . . . . . .
## FO538757.3 . . . . . . . . . . . . . . .
## FO538757.2 . . . . . . . . . 1 . . . . .
## AP006222.2 . . . . . . . . . . . 1 . . .
最後に
いかがだったでしょうか。データセットの結合は思ったよりも簡単だったと思います。実際はバッチエフェクトの補正をしつつ統合させるやり方のほうが多いかもしれませんが、補正しない生データで解析してみるのも重要な解析です。簡単ですので、バッチエフェクト補正の前に結合させただけのデータセット群も解析してみるとなにか新たなことにきづけるかもしれません。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

