
本記事はSeuratのハンズオンの後編になります。前編を取り組んでいない方は先にそちらの方からやるようにしてください。
今回はscRNA-seqのRパッケージであるSeuratのハンズオンに取り組むことで、scRNA-seqを始める前準備を行おうと思います。
本記事はSeuratのハンズオンに則って行いますので使い方の概要を理解するが可能です。ぜひ取り組んでみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
Seuratハンズオンをやってみる(後編)
ハンズオンの準備
前編までに紹介しましたこちらのコードを実行した前提で話を進めます。
実行されていない場合、後半のハンズオンに取り組んでもエラーとなるのでお気をつけください。
# いきなりライブラリ読み込みできなかったのでインストール
install.packages("dplyr")
install.packages("Seurat")
install.packages("patchwork")
library(dplyr)
library(Seurat)
library(patchwork)
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)PCAの前実施
PCAなどの次元削減手法の前に、線形変換による前処理を行います。以下のコマンドを叩いてください。
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)スケーリングされたデータに対してPCAを実行するコマンドは以下になります。
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))各種成分についてこのような出力が得られれば成功です。
PC_ 1
Positive:
CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP
FCER1G, CFD, LGALS1, S100A8, CTSS, LGALS2, SERPINA1, IFITM3, SPI1, CFP
PSAP, IFI30, SAT1, COTL1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD
Negative:
MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27, STK17A, CTSW
CD247, GIMAP5, AQP3, CCL5, SELL, TRAF3IP3, GZMA, MAL, CST7, ITM2A
MYC, GIMAP7, HOPX, BEX2, LDLRAP1, GZMK, ETS1, ZAP70, TNFAIP8, RIC3
PC_ 2
Positive:
CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74
HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB
BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74
Negative:
NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY, B2M, SPON2
CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX
TTC38, APMAP, CTSC, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC
PC_ 3
Positive:
(以下省略)少し見づらいので以下のコマンドで5つに絞って確認します。
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)以下のような出力が得られたら成功です。PC_1〜PC_5まで表示されているか確認してください。
> print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
PC_ 1
Positive: FTL, FTH1, COTL1, CST3, OAZ1
Negative: MALAT1, IL32, LTB, CCL5, CTSW
PC_ 2
Positive: FTL, TYROBP, S100A8, S100A9, FCN1
Negative: ACTG1, STMN1, TUBA1B, TYMS, ZWINT
PC_ 3
Positive: CD74, HLA-DRA, HLA-DPB1, HLA-DQB1, HLA-DQA1
Negative: PPBP, GNG11, SPARC, PF4, AP001189.4
PC_ 4
Positive: CD74, HLA-DQB1, HLA-DQA1, HLA-DRA, HLA-DQA2
Negative: NKG7, GZMA, GNLY, PRF1, FGFBP2
PC_ 5
Positive: ZWINT, KIAA0101, RRM2, HMGB2, AQP3
Negative: NKG7, CD74, HLA-DQA1, GNLY, SPON2ここまででPCAの可視化準備は完了です。ここからは、成分の可視化方法について紹介していきます。
PCAの可視化
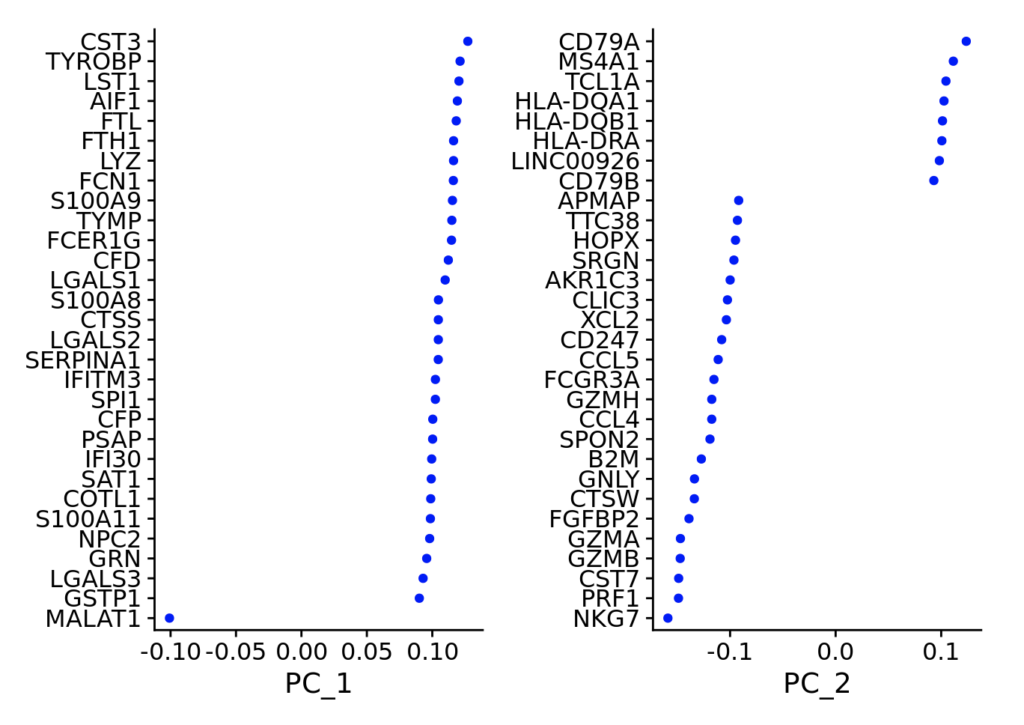
まずは各成分についてどの遺伝子が寄与しているのか確認してみましょう。以下のコマンドを叩いてみてください。
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")寄与率の高い遺伝子から表示ができます。
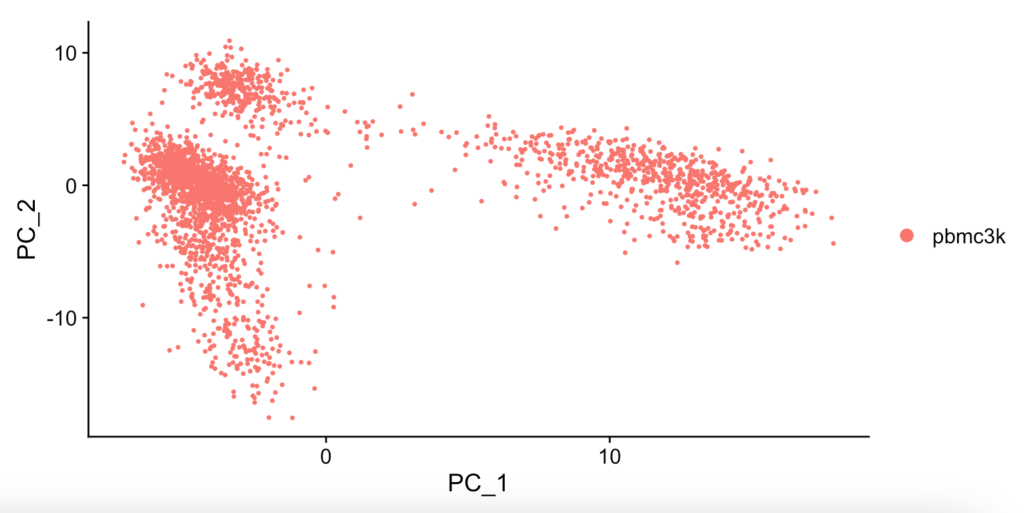
一般的なPC_1とPC_2を用いた散布図を用いてのプロットはDimPlot で行います。
DimPlot(pbmc, reduction = "pca")出力結果は以下になります。
DimHeatmap 関数によって主成分についてheatmap的に調査することができます。表示したい主成分についてdims の引数によって制御します。下記はPC1を表示するコードです。
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)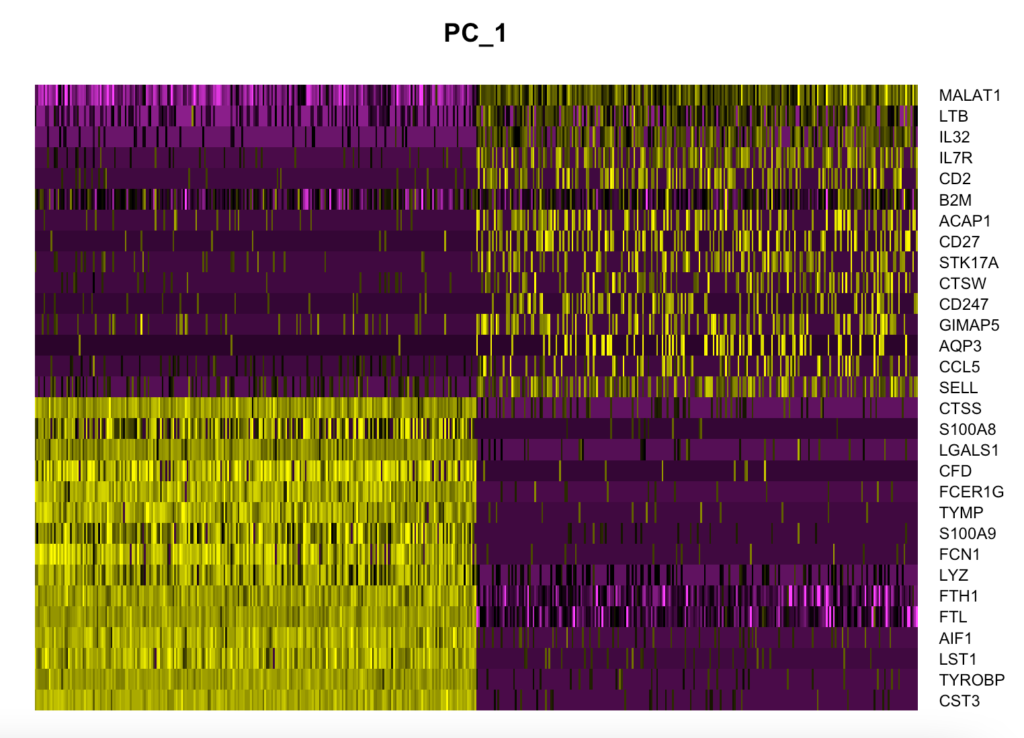
データセットの「次元」を決定する
JackStrawPlotを用いると各主成分のp値について計算し、可視化させることができます。
*こちらのコードは実行に時間がかかります
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
JackStrawPlot(pbmc, dims = 1:15)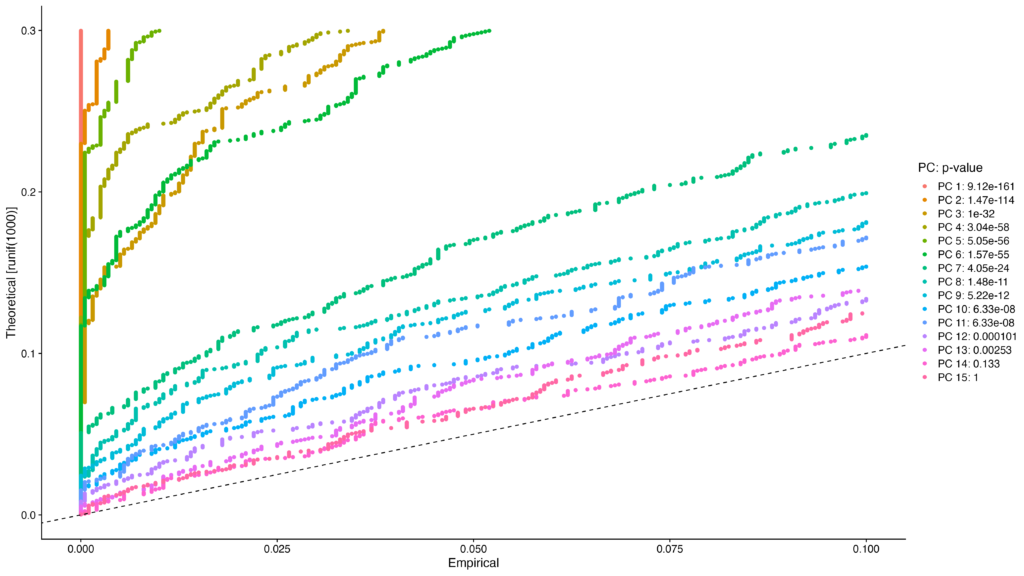
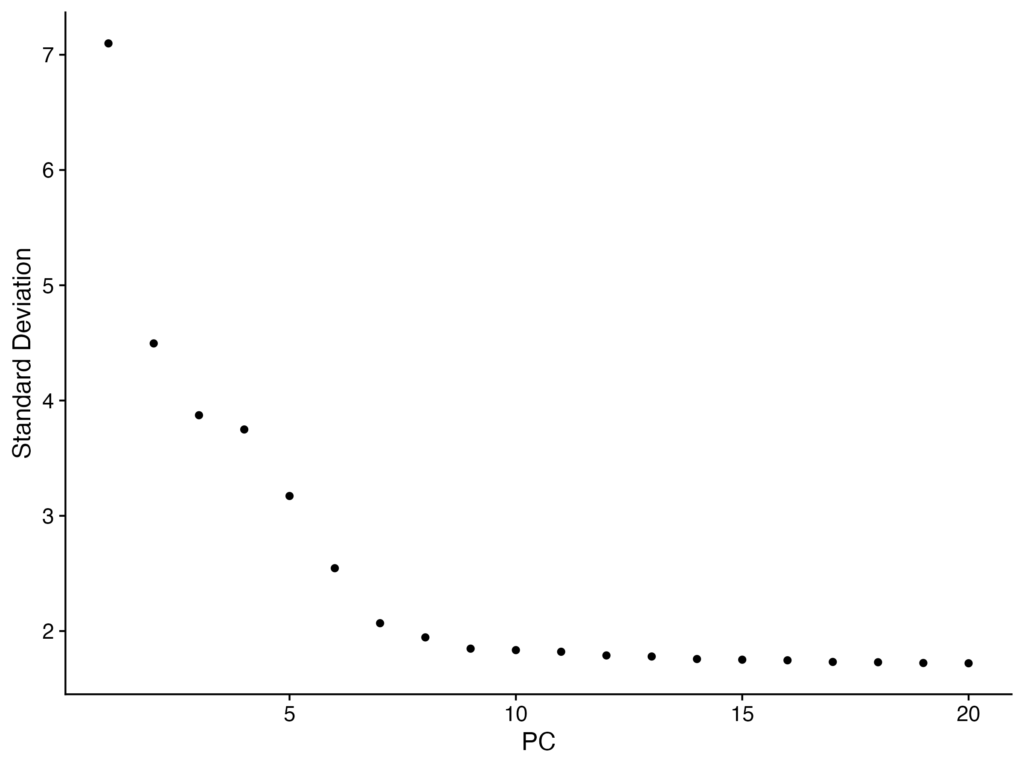
各種成分の割合をランキングするものとしてElbowPlotが可能です。各成分がどれほど寄与しているかを可視化するのに便利です。
ElbowPlot(pbmc)出力は以下になります。PC9-10の周りに「エルボー」が観察され、真のシグナルの大部分が最初の10個のPCで説明されているのがわかります。
セルのクラスタリング(UMAP/tSNE)
いよいよセルのクラスタリングになります。scRNA-seq解析といえば、UMAP/tSNEを用いた非線形次元削減を思い浮かべる人も多いのではないでしょうか。下記コマンドを実行してUMAPを行っていきましょう。
# セルのクラスタリングを行う
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
# UMAPの実行
pbmc <- RunUMAP(pbmc, dims = 1:10)各関数に関しての詳細な解説は以下になります。難しいと思いますので読み飛ばしてもらって大丈夫です。モジュールを最適化してクラスタリングしているんだな程度の理解で大丈夫です。
FindNeighbors関数
PCA空間のユークリッド距離に基づいてKNNグラフを構築し、任意の2つのセル間のエッジの重みを、その局所近傍における共有オーバーラップ(Jaccard類似度)に基づいて洗練させる。入力としてデータセットの先に定義した次元(最初の10個のPC)を取る。
FindClusters関数
セルをクラスタリングするために、次にLouvainアルゴリズム(デフォルト)またはSLMなどのモジュール性最適化技術を適用し、標準モジュール性関数を最適化する目的で、セル間を反復的にグループ化します。下流のクラスタリングの「粒度」を設定するresolutionパラメータを含み、値が大きくなるほどクラスタの数が多くなります。
実行結果をDimPlotで可視化します。
DimPlot(pbmc, reduction = "umap")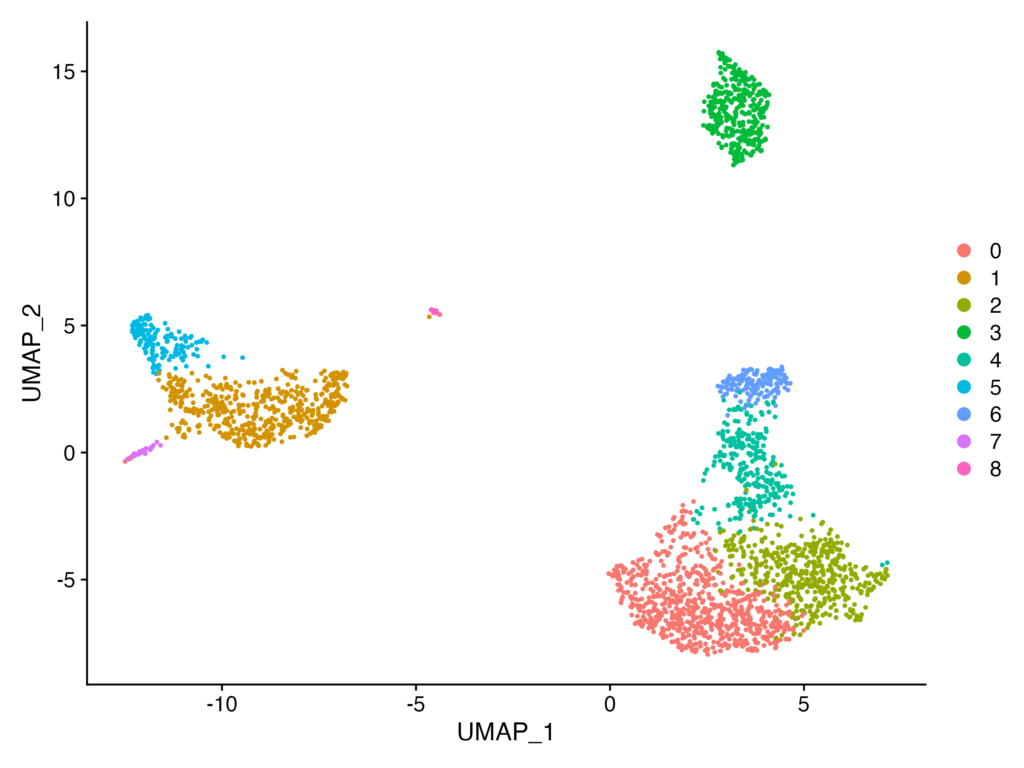
UMAP解析を行うことができました!
ここまでできれば、Seuratを用いたscRNA-seq解析の基礎ができたことになります。ハンズオンではさらなる解析について紹介されていますので、興味がある方は是非チャレンジしてみてください。
もしコマンド通りに叩いてもエラーが出る場合は…
コマンド通りに叩いてもエラーが出る場合は、pbmc に入る値が間違っています。エラーが出ても慌てずに、クオリティチェック→正規化…とコマンドを最初から叩き直してください。順番通りきちんと入力すればエラーなく通ります。
最後に
いかがだったでしょうか。思ったよりもSeuratの使い方も簡単でscRNA-seq解析ができそうだと感じていただけたら幸いです。次は、実際にscRNA-seqデータの探し方を記事に書いて行きたいと思います

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

