
公共データベースのsingle cell rna-seqデータの再解析はできるようになってきたけど、ある程度再解析をしているとbarcode, feature, matrixファイルしか再解析できない現状に満足できなくなってくると思います。
そのため自分でfastqファイルからcountしたいと思うようになるかと思います。この記事では、Cell Rangerで自分でcountできるようになる方法を扱います。
本記事に取り組んで、自分のsingle cell RNA-seq解析の解析幅を広げましょう!
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB, CellBender 0.3.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
Cell Rangerとは?
Cell Rangerは10x Genomics社が開発したソフトウェアパッケージで、シングルセルRNAシーケンシング(scRNA-seq)データの前処理と解析を行うためのツールです。10x GenomicsのChromiumシステムを用いたシングルセルRNA-seq実験から得られるデータの解析に特化しています。下記の図でいうとsequencingした後のデータ処理を行う部分を指しています。Cell Rangerから出力されたOutputはデータの次元削減(t-SNEやPCA)、クラスタリング、差異発現解析など下流の解析へと用いられていきます。
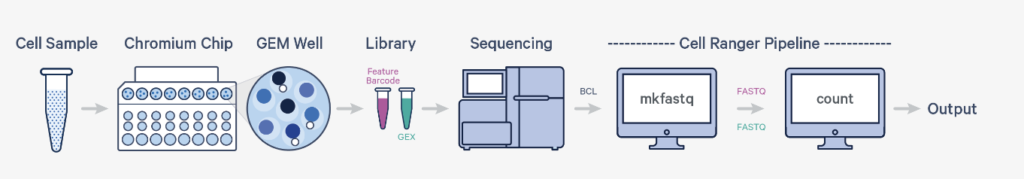
Cell Rangerには以下のような主要なパイプラインがあります。
cellranger mkfastq: 生のBCLファイル(シーケンサーから直接出力されるファイル形式)をFASTQ形式(生物情報解析で一般的に用いられる形式)に変換します。cellranger count: FASTQファイルを入力として、リードのマッピング、フィルタリング、UMIカウント(各細胞・遺伝子の発現量を表すカウント)を行い、細胞ごとの遺伝子発現行列を生成します。cellranger aggr: 複数のcellranger countの結果を統合し、同じ解析空間でこれらのデータを比較可能にします。cellranger reanalyze: すでにcellranger countまたはcellranger aggrで得られた結果を基に、新たなパラメータでクラスタリングや次元削減などの解析を行います。cellranger multi: 異なる種類のシングルセルデータ(RNA-seq, ATAC-seqなど)を同時に処理し、統合解析を行います。
これらのパイプラインを組み合わせることで、生のシーケンスデータから解析結果を得るまでの一連のプロセスを管理することが可能になります。
今回はメインで使うと思われるcellranger mkfastq , cellranger count ,cellranger aggr について、10X genomics社が用意しているハンズオンを参考に紹介したいと思います。
Cell Rangerの実装方法
まずはcell Rangerを扱う環境構築を行います。
大事なことをさらっと言いますが、Cell RangerはLinux OSでしか動かすことができません。これはつまり、我々が普段使っているWindowsやMacでは動かせないということになります。そのためWindowsではUbuntuを入れる、MacではDockerで動かすといった対応が必要になります。(もちろんWindowsでDockerを使っても良いです)
今回は、Docker imageを用いてMac環境でCell Rangerを動かしていこうと思います。Docker Desktopがインストールされている前提で話を勧めます。Windowsの方はこちらの記事の「Ubuntuインストール」の項目を参考にUbuntuをインストールしてください。docker環境を用意しなくて良いwindowsユーザーの方は、そのまあRunning cellranger mkfastqの項目へ飛んでください。
Cell RangerのDocker imageをインストールします。
docker pull litd/docker-cellranger
Docker imageをUIからクリックして起動する。そのままターミナルボタンをおしてターミナル起動してください。
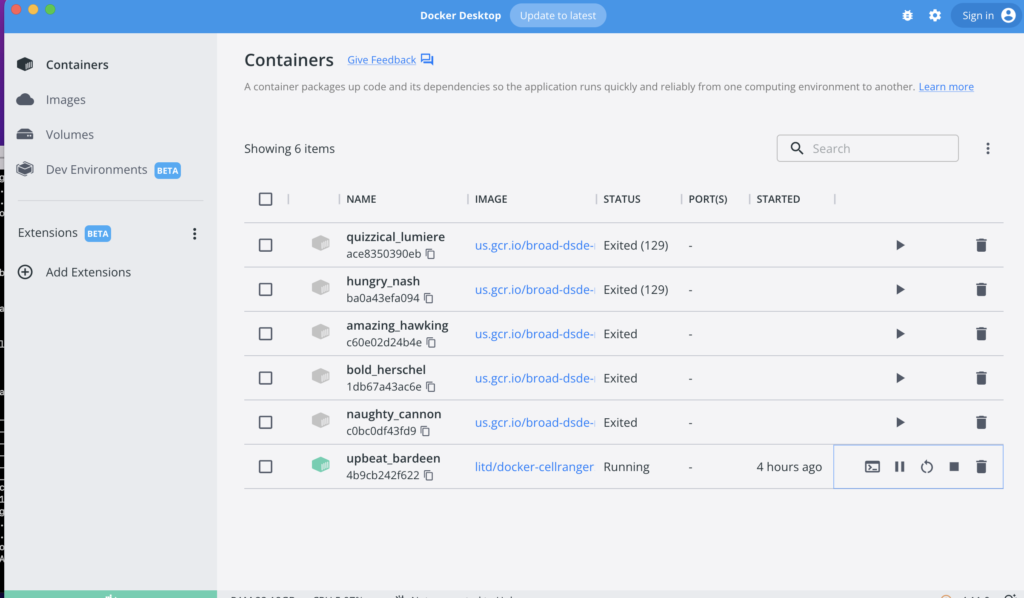
もしUIから起動できない場合はdocker run -it コマンドを使って起動させることも可能ですコンテナIDはUIからコピーできます。
docker run -it [ee0a61935c4f49f03a18ee3a9c1f8c17e4437dda43ca1b5e9d07577565632(コンテナIDの例です)]
起動したら、yardディレクトリを作成してください。基本的な操作はこのyardディレクトリで行います。
mkdir yard
Running cellranger mkfastq
公共データベースにfastqファイルまであれば、こちらのステップはしなくて大丈夫です。そうでない場合は、解析前にmkfastqにてfastqファイルを作成する必要があります。
mkfastqは2つの工程で実行されていきます。
- デモルティプレクシング: デモルティプレクシングとは、一つのシーケンスランに含まれる複数のサンプルを分離するプロセスです。サンプルごとにユニークなバーコードを使用しているため、mkfastqはこれらのバーコードを読み取り、各サンプルのリードを正確に識別します。
- Fastqファイルの生成: デモルティプレクシング後、mkfastqはこの情報を使用してfastqファイルを生成します。Fastqファイルはシーケンスリードとそれらのリードの品質スコアを含むため、後続のシーケンス解析(例えばリードのマッピング、遺伝子カウントなど)に必要な入力となります。
では実際に、mkfastqを実行してみましょう。run_cellranger_mkfastq ファイルを作成して、中に入ってください。
mkdir /yard/run_cellranger_mkfastq
cd /yard/run_cellranger_mkfastqサンプルファイルをダウンロードして、解凍します。
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-1.2.0.tar.gz
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-simple-1.2.0.csv
tar -zxvf cellranger-tiny-bcl-1.2.0.tar.gz解凍が成功すると、以下のようなファイル郡が出力されると思います。
cellranger-tiny-bcl-1.2.0/
cellranger-tiny-bcl-1.2.0/Data/
cellranger-tiny-bcl-1.2.0/Data/Intensities/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C25.1/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C25.1/s_1_1101.bcl.gz
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C1.1/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C1.1/s_1_1101.bcl.gz
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C2.1/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C2.1/s_1_1101.bcl.gz
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C3.1/
cellranger-tiny-bcl-1.2.0/Data/Intensities/BaseCalls/L001/C3.1/s_1_1101.bcl.gz
.
.
.
下記コマンドでmkfastqを実行します。
cellranger mkfastq --id=tutorial_walk_through \\
--run=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-1.2.0 \\
--csv=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-simple-1.2.0.csv
以下のような出力が出ればが作成されれば成功です!
sh-4.4# cellranger mkfastq --id=tutorial_walk_through \\
> --run=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-1.2.0 \\
> --csv=/yard/run_cellranger_mkfastq/cellranger-tiny-bcl-simple-1.2.0.csv
/opt/cellranger-7.1.0/bin
cellranger mkfastq (cellranger-7.1.0)
Copyright (c) 2021 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Martian Runtime - v4.0.10
2023-06-11 11:57:52 [runtime] Reattaching in local mode.
Serving UI at <http://4b9cb242f622:39525?auth=TKb2F_7AUK6OGai2jN2JBiMtSEysYDXQV3OIbXZURdo>
Outputs:
- FASTQ output folder: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/fastq_path
- Interop output folder: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/interop_path
- Input samplesheet: /yard/run_cellranger_mkfastq/tutorial_walk_through/outs/input_samplesheet.csv
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
2023-06-11 11:57:58 Shutting down.
Saving pipestance info to "tutorial_walk_through/tutorial_walk_through.mri.tgz"
作成されたfastqファイルは、/yard/run_cellranger_mkfastq/tutorial_walk_through/outs/fastq_path/H35KCBCXY/test_sample に格納されていますので以下のコマンドで見てみましょう。
cd tutorial_walk_through/outs/fastq_path/H35KCBCXY/test_sample
ls
以下のようにfastqファイルが確認できると思います。
sh-4.4# ls
test_sample_S1_L001_I1_001.fastq.gz test_sample_S1_L001_R1_001.fastq.gz test_sample_S1_L001_R2_001.fastq.gz
Running cellranger count
fastqファイルから遺伝子発現のカウントを得るためにcellranger countを実行します。これはCell Rangerパイプラインの主要なステップで、リードのマッピング、フィルタリング、バーコードのカウント、UMI(Unique Molecular Identifier)のカウントなど、一連の解析が行われます。
まず、run_cellranger_countディレクトリを作成し、移動します
mkdir /yard/run_cellranger_count
cd /yard/run_cellranger_count
サンプルファイルをダウンロードして解凍してください。
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_fastqs.tar
tar -xvf pbmc_1k_v3_fastqs.tar
cellranger countコマンドにはレファレンスゲノムが必要です。サンプルと同じようにダウンロードしてください。
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
tar -zxvf refdata-gex-GRCh38-2020-A.tar.gz
cellranger countコマンドを実行します。このコマンドでは、解析対象のfastqファイルの場所、参照ゲノム、出力ディレクトリの名前などを指定します。このコマンドの実行には数時間かかります。
cellranger count --id=run_count_1kpbmcs \\
--fastqs=/yard/run_cellranger_count/pbmc_1k_v3_fastqs \\
--sample=pbmc_1k_v3 \\
--transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A
引数の詳細は次の通りです:
-id=run_count_1kpbmcs: 出力ディレクトリの名前を指定します。この場合、出力フォルダの名前は “run_count_1kpbmcs” となります。--fastqs=/yard/run_cellranger_count/pbmc_1k_v3_fastqs: 入力FASTQファイルが格納されているディレクトリのパスを指定します。この場合、FASTQファイルは “/mnt/home/user.name/yard/run_cellranger_count/pbmc_1k_v3_fastqs” ディレクトリに存在します。--sample=pbmc_1k_v3: サンプル名を指定します。FASTQファイルの名前にこのサンプル名が含まれている必要があります。この例では、FASTQファイルの名前に “pbmc_1k_v3” が含まれていると想定されます。--transcriptome=/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A: 使用するトランスクリプトームの参照データのパスを指定します。この場合、参照データは “/mnt/home/user.name/yard/run_cellranger_count/refdata-gex-GRCh38-2020-A” ディレクトリに存在します。
完了すると、/yard/run_cellranger_count/tutorial_count/outsディレクトリに結果が格納されます。出力ファイルを確認してみましょう。
cd /yard/run_cellranger_count/tutorial_count/outs
ls
出力結果は、各サンプルや解析条件により異なりますが、一般的に以下のようなファイルが出力されるはずです。
analysis filtered_feature_bc_matrix.h5 metrics_summary.csv
cloupe.cloupe filtered_feature_bc_matrix molecule_info.h5
filtered_feature_bc_matrix possorted_genome_bam.bam web_summary.html
このディレクトリには、遺伝子ごとのUMIカウントマトリクス(filtered_feature_bc_matrix)、アライメントの結果(possorted_genome_bam.bam)、クラスタリングと次元削減の結果(cloupe.cloupe)などが含まれます。
これでcellranger countの実行が完了です。
Running cellranger aggr
cellranger aggrは複数のcellranger countの出力(つまり、複数のサンプルや実験からのシングルセルRNA-seqデータ)を組み合わせるためのパイプラインです。このパイプラインは各データセットを正規化し、それらを一つの表現行列に結合します。これにより、複数のサンプルや条件をまたいだ解析が可能となります。cellranger countをして必要であれば実行してください。
まずはrun_cellranger_aggrディレクトリを作成してください。
mkdir run_cellranger_aggr
cd run_cellranger_aggr
サンプルファイルをダウンロードして解凍してください。
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_molecule_info.h5
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_molecule_info.h5
cellranger aggrにはサンプルまでのファイルパスを示したcsvを作成する必要があります。
以下に、2つのサンプル(sample1とsample2)を集約するための入力ファイル(libraries.csv)の例を示します:
viコマンドでpbmc_aggr.csvを作っていきます。
vi pbmc_aggr.csv
pbmc_1k_v3_molecule_info.h5 とpbmc_10k_v3_molecule_info.h5のファイルパスを示したpbmc_aggr.csvを作成します。
sample_id,molecule_h5
1k_pbmcs,path/to/run_cellranger_aggr/pbmc_1k_v3_molecule_info.h5
10k_pbmcs,path/to/run_cellranger_aggr/pbmc_10k_v3_molecule_info.h5
そして、このCSVファイルを用いてcellranger aggrコマンドを実行します
cellranger aggr --id=1k_10k_pbmc_aggr --csv=pbmc_aggr.csv
このコマンドにより、入力された各サンプルのデータが統合され、一つのカウントマトリクスとして出力されます。出力ファイルは/yard/run_cellranger_aggr/tutorial_aggr/outsディレクトリに格納されます。出力ファイルを確認してみましょう。
cd /yard/run_cellranger_aggr/tutorial_aggr/outs
ls
出力結果は、入力データや解析条件により異なりますが、一般的に以下のようなファイルが出力されるはずです。
analysis filtered_matrices_mex raw_feature_bc_matrix
cloupe.cloupe raw_feature_bc_matrix.h5 web_summary.htmlこれでcellranger aggrの実行が完了しました。これにより得られたデータを解析してみましょう。
最後に
Cell Rangerを用いてfastqファイルからのカウント方法を学びました。公共データベースの再解析だけでなく、自ら初歩からデータを解析することは一見難しそうに見えますが、独自の探究と発見のドアを開けるための重要なステップです。是非挑戦してみてください。
参考

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

