
scRNA-seqデータはクオリティコントロール(QC)をすると聞いたことがあるかもしれませんが詳しく理解ができていない人もいるのではないでしょうか。
ここではSingle Cell RNA-seqサンプルをCellBenderを使ってノイズを除去する方法を学びます。
これまで漠然としていたscRNA-seqのクオリティコントロールについてわかるようになります。ぜひ挑戦してみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB, CellBender 0.3.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
CellBenderとは?
CellBenderには解説ページが用意されているので、今回はこちらをもとに解説していきます。
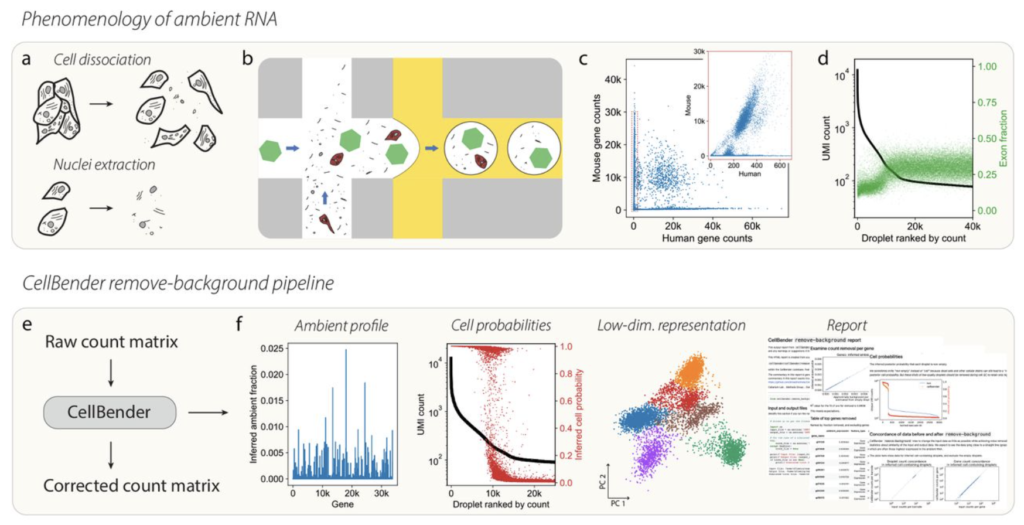
CellBenderとは、scRNA-seqデータにしばしば存在する実験的なノイズやアーチファクト(背景ノイズ、ドロップアウト、空のドロプレットなど)を処理するためのソフトウェアです。
- 空のドロプレットの識別:ドロップシーケンシングでは、細胞がキャプチャされなかったドロプレットでもRNAが検出されることがあります。これらは「空のドロプレット」と呼ばれ、しばしばノイズとなります。CellBenderは、これらの空のドロプレットを効率的に識別し、データから除去します。
- ドロップアウトの補正:シングルセルRNAシーキングデータでは、特定の遺伝子が検出されない(ドロップアウト)ことがしばしばあります。CellBenderはベイズ統計的な手法を用いて、これらのドロップアウトを補正します。
- 深層学習に基づくモデル:CellBenderは深層学習(特に変分自己符号化器:VAE)に基づくモデルを使用します。これにより、高次元のscRNA-seqデータの複雑なパターンを効率的に学習し、ノイズを除去します。
https://www.biorxiv.org/content/10.1101/791699v2.full
CellBenderでは、バーコード、フィーチャー、マトリックスファイルではなく、hdf5ファイルを使って解析します。バーコード、フィーチャー、マトリックスファイルとhdf5ファイルの違いを軽く触れておきます。
バーコード、フィーチャー、マトリックスの3つのファイル形式は、通常、10x GenomicsのようなscRNA-seqプラットフォームでよく使われます。各ファイルは以下の情報を含んでいます:
- バーコードファイル:個々の細胞を識別するための一意のバーコードのリスト
- フィーチャー(遺伝子)ファイル:測定された全遺伝子のリスト
- マトリックスファイル:各細胞での各遺伝子の発現レベルを含む疎な行列
一方、HDF5ファイルは、これらの情報をすべて1つのファイルに統合します。HDF5は階層的データ形式で、大量のデータを高効率に格納できます。
この2つの形式の主な違いは、読み込みと解析に必要な手順の数です。バーコード、フィーチャー、マトリックスの3つのファイルを使用する場合、各ファイルを個別に読み込み、それらを一緒に解析するためのマトリックスを組み立てる必要があります。しかし、HDF5形式では、すべての情報が1つのファイルに含まれているため、単一の操作でファイルを読み込むことができます。
CellBenderにおける注意点とヒント
“remove-background”は、空の液滴(細胞がない液滴)に含まれる一定量の背景RNAを除去します。これは「ambient plateau」と呼ばれるフェノメノンを示しています。
Exhibit Aのように、特定のデータセットでは空の液滴のUMIカウントが非常に少ない場合、バックグラウンドRNAはほとんど存在せず、この機能はあまり効果が見られないかもしれません。
Exhibit Bのように、UMIカウントが50、100、数百程度の空の液滴が視覚的に確認できるデータセットについては、”remove-background”機能によりデータセットが大幅に改善されると期待できます。
Exhibit Cのように、背景RNAが非常に多くて「空の液滴のプラトー」が視覚的に確認できないデータセットについては、”remove-background”の実行は困難です。この場合は実験から再実行するのが推奨されています。
Dockerのインストール
まずはDockerをインストールしていきます。
Dockerのインストール
Dockerを用いるのでDockerをインストールしていきます。DockerのインストールはMac版とWindows版で対応手順が異なるため分けて記載します。
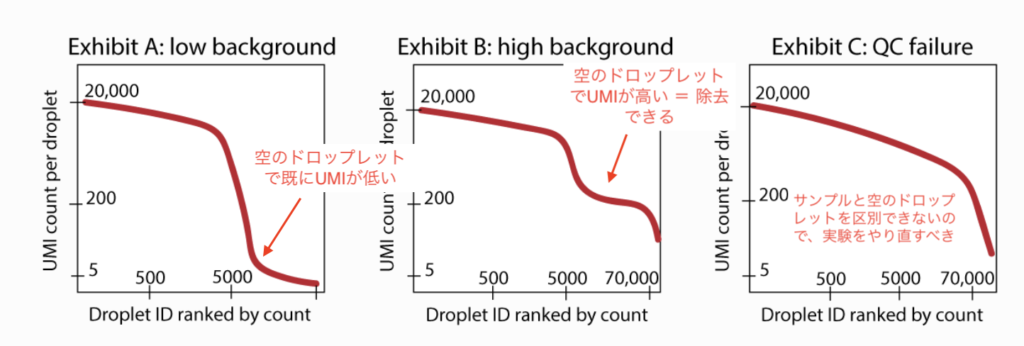
【Macの場合】
Mac版はIntel chipとApple chip(M1系)に分かれているので注意してください。Macの方は以下のURLよりダウンロードしてください。(ダウンロードに5分程かかります。)
ダウンロードが終わったら、指示に従ってインストールを完了させてください。インストールが終わったらDockerのアイコンが出来上がっていると思いますのでクリックして起動しておいてください。
【Windowsの場合】
Windowsユーザーの方がDockerを使用するためには、先にWSL2上へのUbuntuのインストールをする必要があります。順を追って対応していきましょう。
Ubuntuのインストール
Linux用Windowsサブシステムを有効にしていきます。
- PowerShellを「管理者として実行」する
- 下記コマンドで、LinuxのディストリビューションであるUbuntuをインストール
wsl --install -d Ubuntu
!https://zenn.dev/images/copy-icon.svg
下記のような出力がされます。
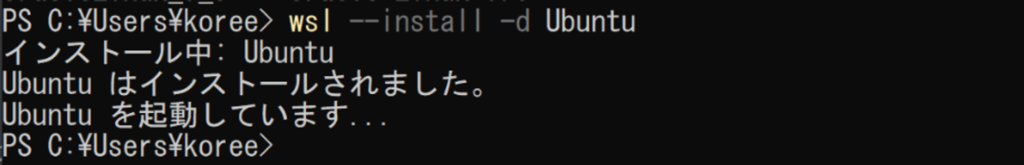
インストールが完了して、Ubuntuアイコンをクリックしてみると下記のようなエラーになり起動できないかと思います。
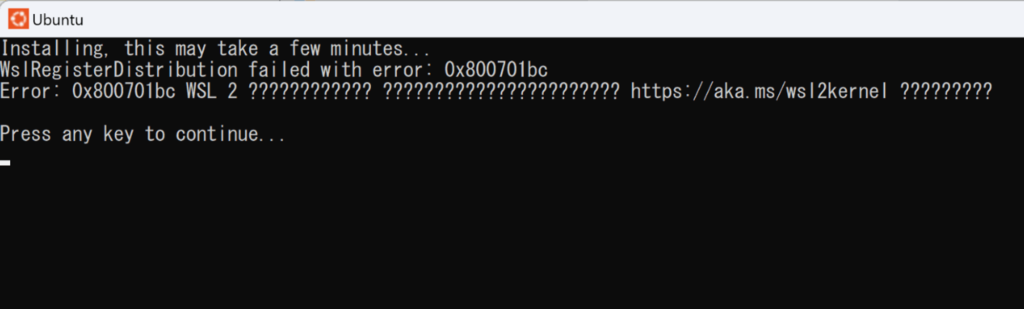
これは、Linuxカーネル更新プログラムパッケージをダウンロードすることで解決します。Microsoftの以前のバージョンの WSL の手動インストール手順 (下記の画像)のところから最新版のパッケージをダウンロードしてください。
最新版のパッケージをダウンロードしたらインストールを進め、Finishを押してください。
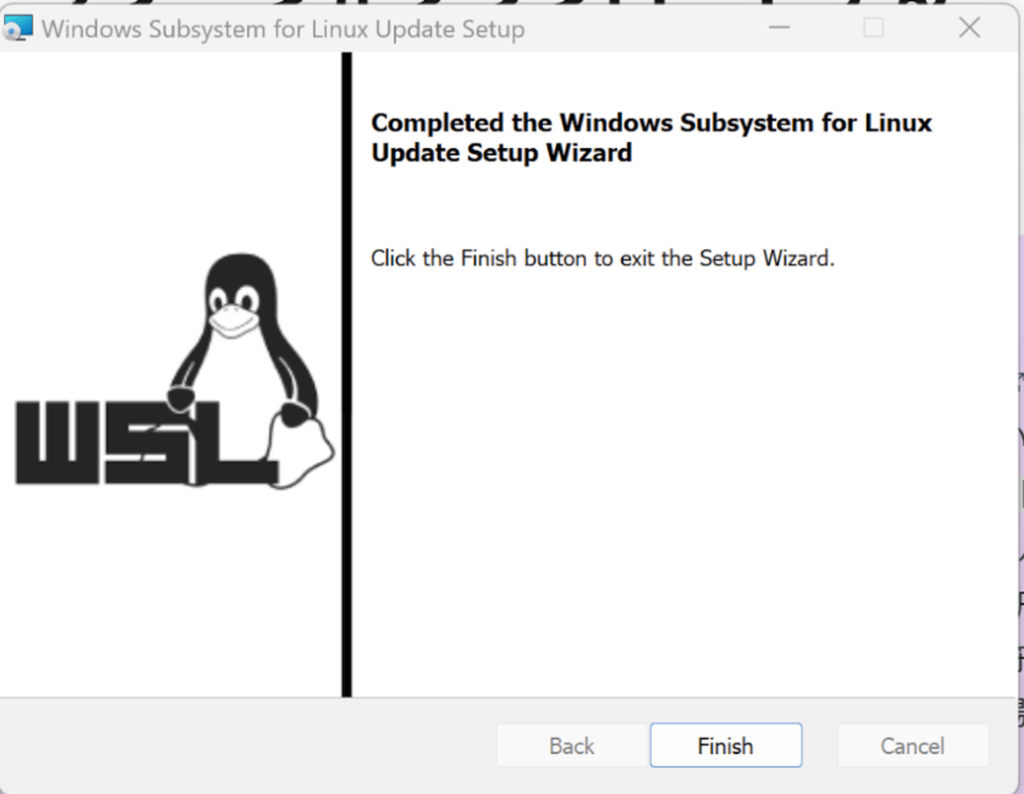
これでUbuntuが起動できるようになります。UNIX usernameとpasswordを決めると今度は起動できると思います。
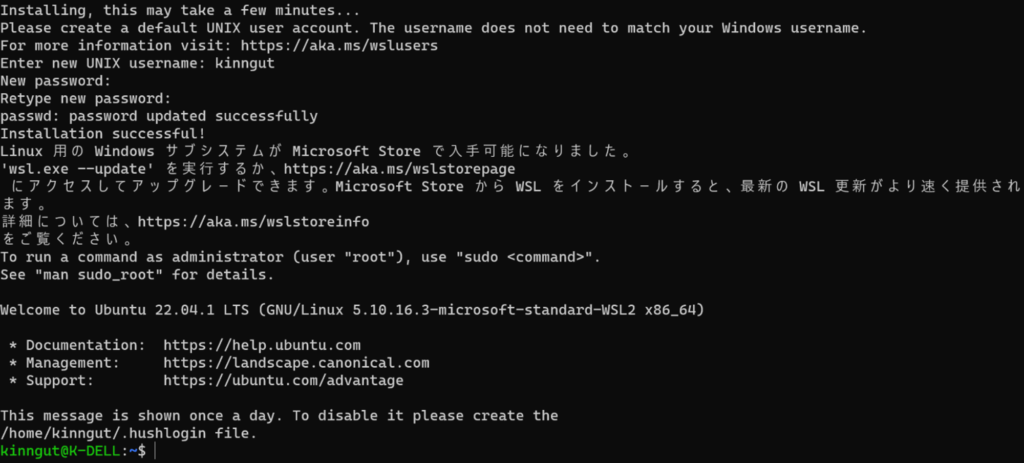
Ubuntuのインストールが確認できましたらWindows版のDockerをインストールしてください。
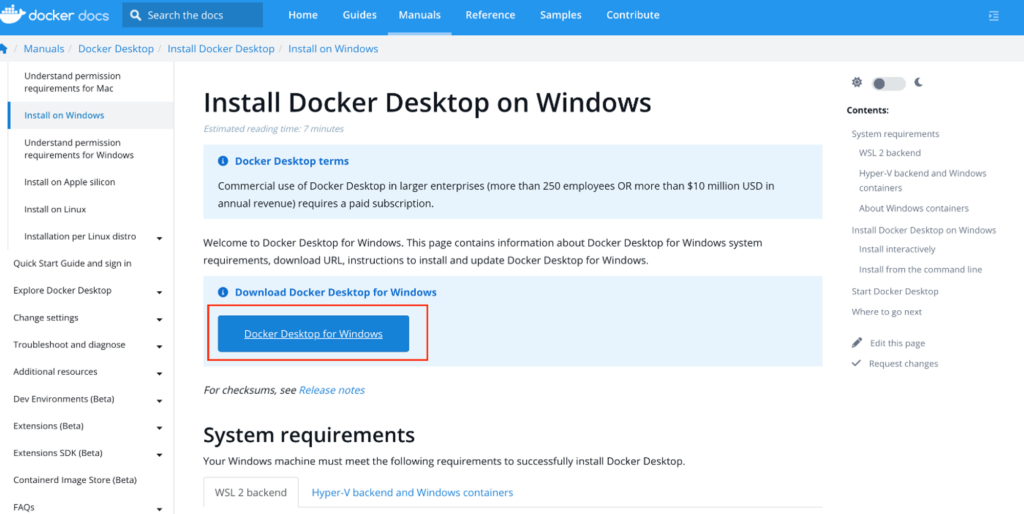
ダウンロードが終わったら、指示に従ってインストールを完了させてください。インストールが終わったらDockerのアイコンが出来上がっていると思いますのでクリックして起動しておいてください。
CellBenderを実行する方法
前準備が長くなりましたが、いよいよCellBenderを実行していきます。CellBenderの公式ドキュメントを参考にします。
Docker imageをインストールします。
docker pull us.gcr.io/broad-dsde-methods/cellbender:latest
Dockerを起動させます。
docker run -it us.gcr.io/broad-dsde-methods/cellbender:latest /bin/bash
cellbender/examples/remove_background階層に移動し、
cd cellbender/examples/remove_background
ここでpython コマンドを叩き、generate_tiny_10x_pbmc.py を起動させるのですが、筆者の環境ではエラーが出ました。
root@ba0a43efa094:/cellbender/examples/remove_background# python generate_tiny_10x_pbmc.py
Downloading pbmc4k (CellRanger 2.1.0, v2 Chemistry)...
Could not retrieve pbmc4k (CellRanger 2.1.0, v2 Chemistry) -- cannot continue!
これを回避していきます。wgetコマンドでサンプルのダウンロードを行います。
wget http://cf.10xgenomics.com/samples/cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz
pbmc4k_raw_gene_bc_matrices.tar.gzファイルがダウンロードされます。
generate_tiny_10x_pbmc.py を編集していきます。まずは、vimをダウンロードします。
# Mac
apt-get update
apt-get install vim
# Windows
sudo apt update
sudo apt install vim
以上の操作でviコマンドが使えるようになったと思います。実際に下記を実行して行きましょう。
vi generate_tiny_10x_pbmc.py
vimモードでは「i」を打つと、INSERTモードとなり編集が可能となります。
下記を参考に編集を行ってください。編集後はescキーを押してInsertモードを解除後、「:wq」と打ち込んでエンターを押すと保存ができます。タブが崩れないように注意してください。
import urllib.request #このライブラリを消す
import requests #追記
import sys
import tarfile
import os
import numpy as np
from scipy.io import mmread, mmwrite
import pandas as pd
import operator
import shutil
dataset_name = "pbmc4k (CellRanger 2.1.0, v2 Chemistry)"
dataset_url = "<http://cf.10xgenomics.com/samples/cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz>"
dataset_local_filename = "pbmc4k_raw_gene_bc_matrices.tar.gz"
expected_cells = 4000
num_cell_barcodes_to_keep = 500
num_empty_barcodes_to_keep = 50_000
num_genes_to_keep = 100
random_seed = 1984
rng = np.random.RandomState(random_seed)
#------ここから------------------------------------------------
try:
print(f"Downloading {dataset_name}...")
urllib.request.urlretrieve(dataset_url, dataset_local_filename)
except IOError:
print(f"Could not retrieve {dataset_name} -- cannot continue!")
sys.exit()
#------ここまでを削除---------------------------------------------
# 以下を新しく追記
try:
print(f"Downloading {dataset_name}...")
response = requests.get(dataset_url, stream=True)
response.raise_for_status() # Raise an exception in case of HTTP errors
with open(dataset_local_filename, 'wb') as f:
for chunk in response.iter_content(chunk_size=1024):
if chunk: # Filter out keep-alive new chunks
f.write(chunk)
except requests.exceptions.RequestException as err:
print(f"Could not retrieve {dataset_name} -- cannot continue! Error: {err}")
sys.exit()
修正が終わったら実行していきます。
python generate_tiny_10x_pbmc.py
その後に以下のコマンドを叩きます。実行には6~12時間ほどかかります。GPUが使える方は、-cuda フラグを用いると実行時間が短縮できます。
cellbender remove-background \\
--input ./tiny_raw_gene_bc_matrices/GRCh38 \\
--output ./tiny_10x_pbmc.h5 \\
--expected-cells 500 \\
--total-droplets-included 5000
各オプションの説明は以下の通りです。
-input ./tiny_raw_gene_bc_matrices/GRCh38:入力データのパスを指定します。ここでは、現在のディレクトリにある “tiny_raw_gene_bc_matrices/GRCh38” フォルダが指定されています。-output ./tiny_10x_pbmc.h5:バックグラウンドノイズが除去された後のデータを保存するパスを指定します。ここでは、現在のディレクトリに “tiny_10x_pbmc.h5” という名前のファイルが作成されます。-expected-cells 500:予想される実際の細胞の数を指定します。これはアルゴリズムがデータを解釈するための重要なパラメーターであり、通常は実験から得られる情報に基づいて設定されます。ここでは、500個の細胞が期待されています。-total-droplets-included 5000:解析に含めるドロップレット(細胞を含む可能性のある小さなビーズ)の総数を指定します。ここでは、5000個のドロップレットが含まれることが期待されています。
以下のようになれば成功しています!
root@ba0a43efa094:/cellbender/examples/remove_background# ls
generate_tiny_10x_pbmc.py tiny_10x_pbmc.h5 tiny_10x_pbmc.log tiny_10x_pbmc.pdf tiny_10x_pbmc_cell_barcodes.csv tiny_10x_pbmc_filtered.h5
以下に各出力ファイルの説明をします:
tiny_10x_pbmc.h5: これはHDF5形式のファイルで、推論手順の詳細な出力を含んでいます。これには、正規化された環境トランスクリプトの豊富さ、各ドロップレットの汚染度合い、バックグラウンド補正後の遺伝子発現の低次元埋め込み、およびバックグラウンド補正後のカウント行列(CSC疎行列形式)などが含まれます。tiny_10x_pbmc_filtered.h5: 上記と同じ情報を含みますが、こちらは後方の細胞確率が0.5を超えるドロップレットのみを含んでいます。tiny_10x_pbmc_cell_barcodes.csv: 後方の細胞確率が0.5を超えるバーコードのリストです。tiny_10x_pbmc.pdf: トレーニング中の損失関数の進化、UMIプロットと後方細胞確率のランク付け、細胞を含むドロップレットの発現の潜在的な埋め込みの2次元PCA散布図を示す結果のPDFサマリーです。UMIランクが約500を超えると細胞確率が急速に下がることに注目してください。
生成されたファイルを解析しますので、ローカルにコピーします。Docker DesktopからコンテナIDをコピーしてください。
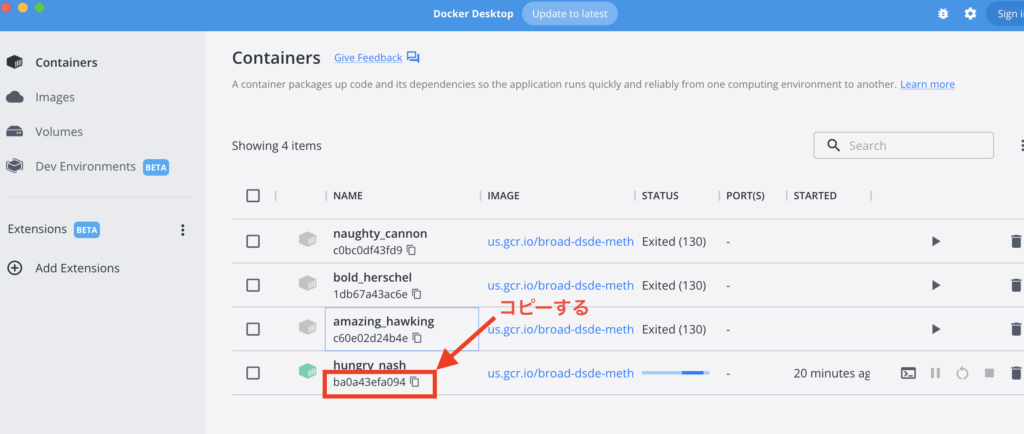
別でターミナルを開いて先程コピーしたIDを使って、ファイルをコピーします(Dockerのターミナルではなく新しいターミナルを使います)
docker container cp [コピーしたID]:/cellbender/examples /Users/hogeuser/Desktop/
Desktopにexamplesファイルだけダウンロードできたかと思います。ここで重要なのは、tiny_10x_pbmc_filtered.h5 ファイルができていることを確認することです。
tiny_10x_pbmc_filtered.h5 ファイルをSeuratで解析していきます。Read10X_h5 関数を使いたいので、hdf5r ライブラリを一緒にインストールして使っていきます。
# load Seurat (version 3) library
library(Seurat)
install.packages("hdf5r")
library(hdf5r)
# load data from the filtered h5 file
data.file <- 'tiny_10x_obj_filtered.h5'
data.data <- Read10X_h5(filename = data.file, use.names = TRUE)
# Initialize the Seurat object with the raw (non-normalized data).
obj <- CreateSeuratObject(counts = data.data)
# Data normalization
obj <- NormalizeData(obj)
# Find variable features
obj <- FindVariableFeatures(obj)
# Scale data
obj <- ScaleData(obj)
obj <- RunPCA(obj, features = VariableFeatures(object = obj))
# Cluster the cells
obj <- FindNeighbors(obj)
obj <- FindClusters(obj)
# Run non-linear dimension reduction (UMAP/tSNE)
obj <- RunUMAP(obj, dims = 1:10)
# Plot the UMAP
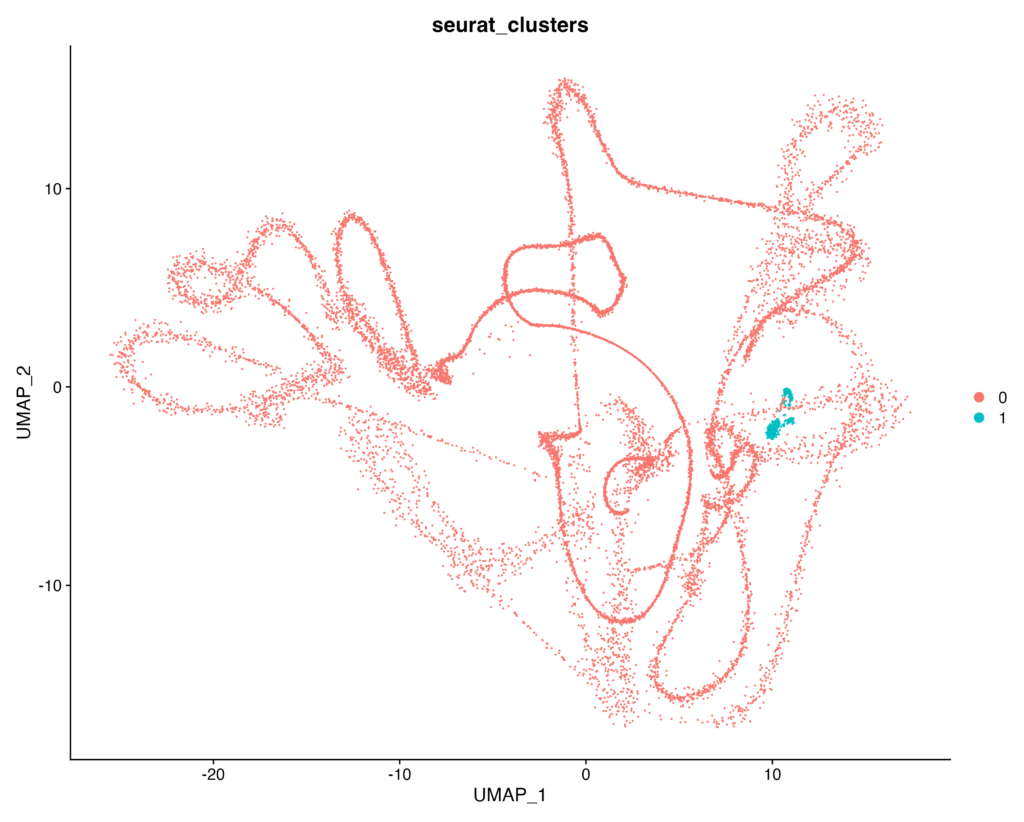
DimPlot(obj, group.by = "seurat_clusters")
以下のようなプロットが生成されれば成功です!
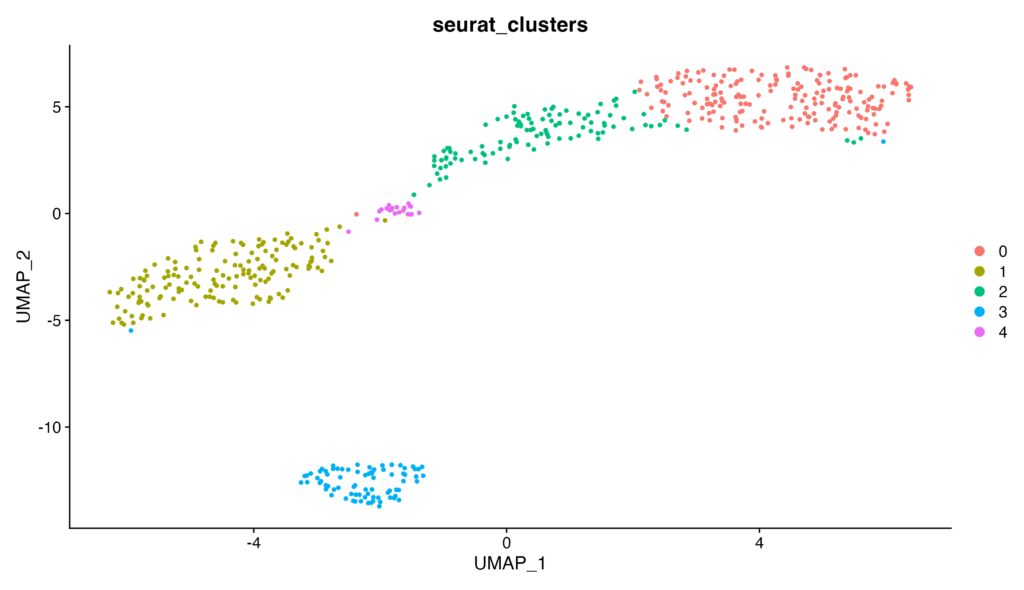
解説
tiny_10x_pbmc.h5とtiny_10x_pbmc_filtered.h5 をそれぞれseuratオブジェクトにし、サンプル数を出力してみると、
# load data from the filtered h5 file
data.data_filtered <- Read10X_h5(filename = 'tiny_10x_pbmc_filtered.h5', use.names = TRUE)
data.data <- Read10X_h5(filename = 'tiny_10x_pbmc.h5', use.names = TRUE)
# Initialize the Seurat object with the raw (non-normalized data).
obj_filterd <- CreateSeuratObject(counts = data.data_filtered)
obj <- CreateSeuratObject(counts = data.data)
# ---出力結果---
> obj_filterd
An object of class Seurat
100 features across 558 samples within 1 assay
Active assay: RNA (100 features, 0 variable features)
> obj
An object of class Seurat
100 features across 14650 samples within 1 assay
Active assay: RNA (100 features, 0 variable features)
となり、fileter前は14650細胞あったが、filter後は558細胞になっていることが分かります。ほとんどがカットされていますね。tiny_10x_pbmc.pdf の方も見てみましょう。
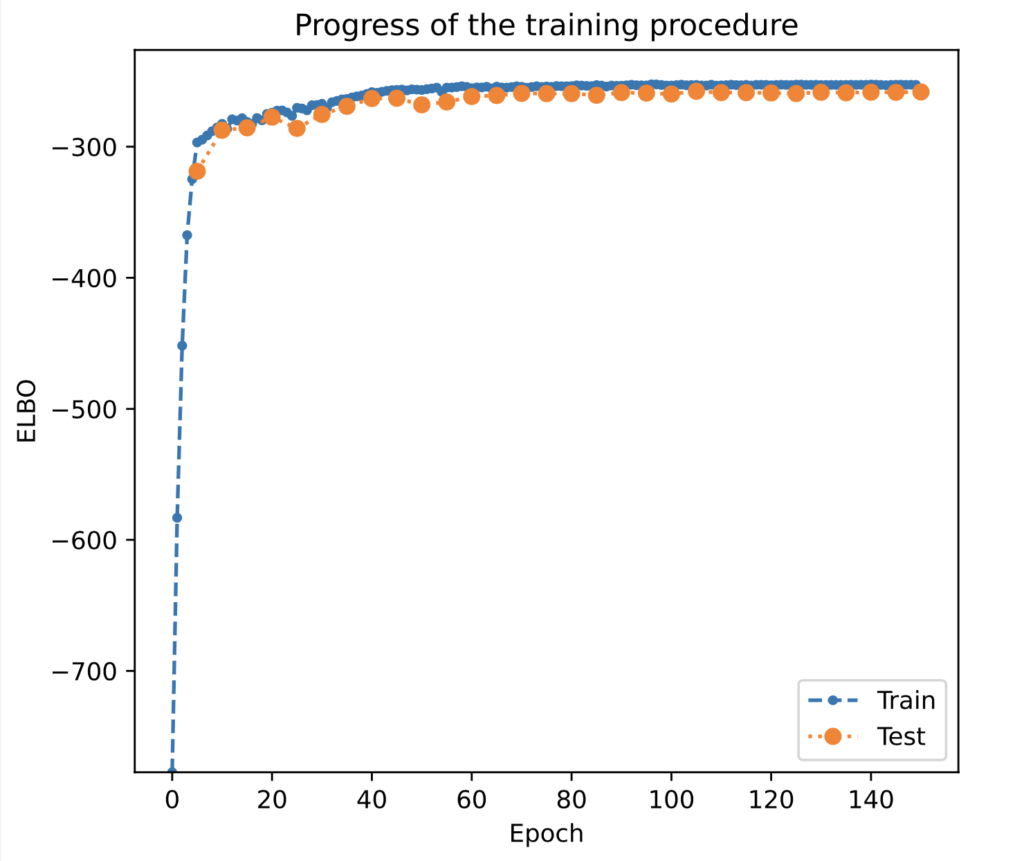
「ELBO」は “Evidence Lower Bound” の略で、ベイズ統計学における最適化目標関数の一つです。ELBOが増加しない、あるいは大きく下降している場合、それは学習率が高すぎて収束していない可能性を示します。
「Epoch」は、訓練データ全体が学習アルゴリズムに何回通過したかを示す指標です。
ELBOの値が単調に増加するのではなく、大きく下降するようであれば(ノイズは別として)、学習率を2倍下げてみてください。
もしELBOの値が適度に安定した値に収束して行かなければ、--epochsのオプションでエポック数を増やして再実行することがおすすめされています。 目安としては、300を超えないようにするべきだと述べられています。
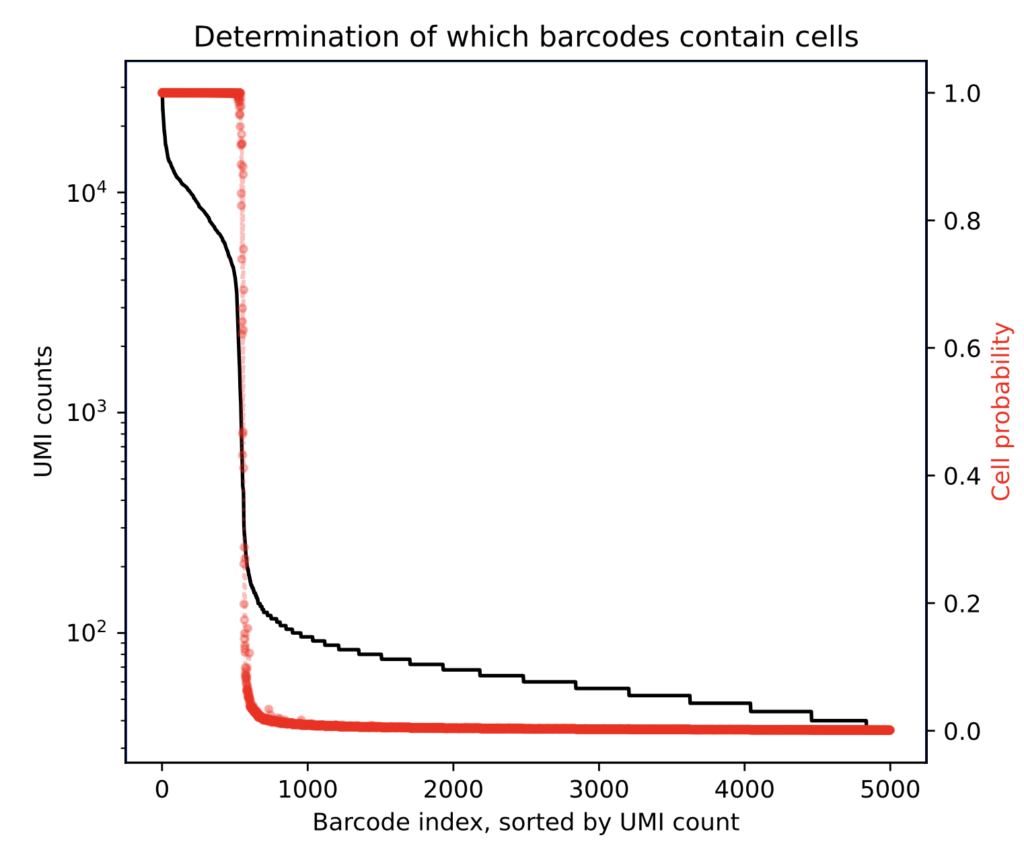
次の図では、サンプルカウントが500を超えたあたりで補正後のCell probability(赤のライン)が0の方へ分類されているのが分かります。これにより、サンプルと空のドロップレットの区別が明確になりました。
こちらは遺伝子発現をコード化する潜在変数についてPCAを行っています。この段階でいくつかクラスターが見られると良い傾向です。クラスタリングの欠如は、データセットが1つの細胞タイプしか持っていないことなどQC問題を示している可能性があります。
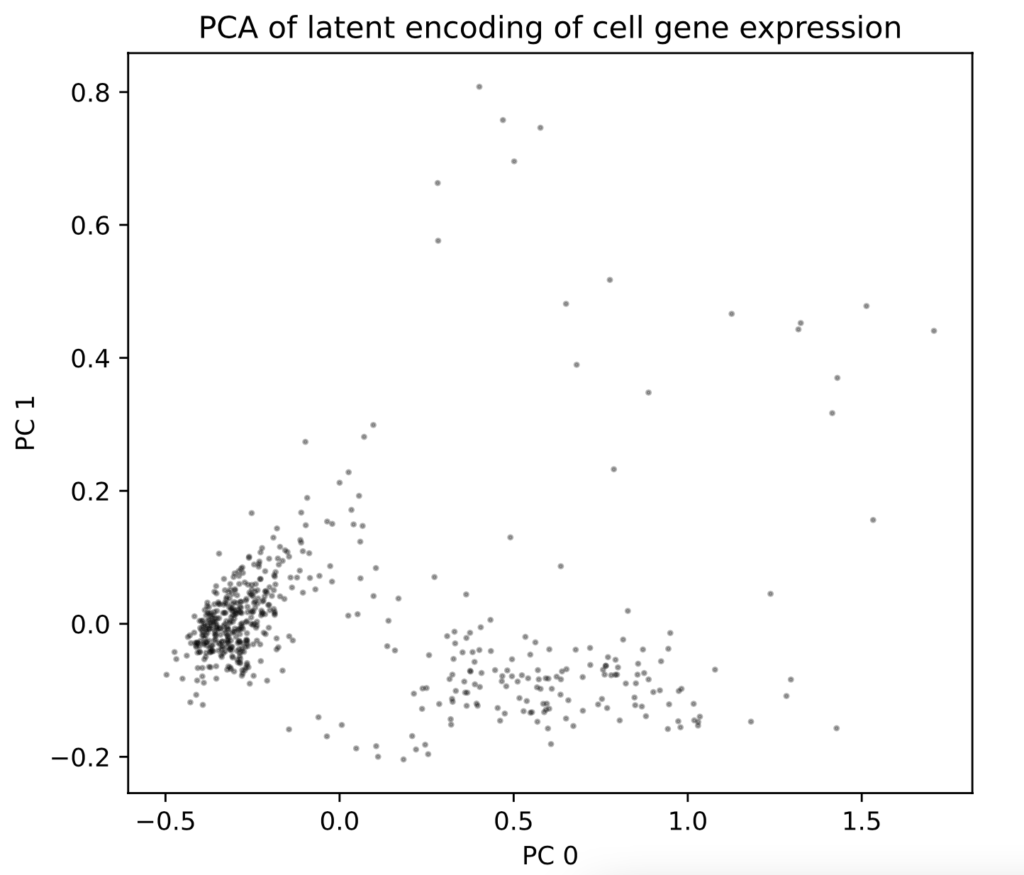
試しに tiny_10x_pbmc.h5を解析してUMAPプロットまで持っていくと、クラスターをなしておらずきちんと解析ができません。QCがいかに重要かが分かりますね。
パラメータを設定する際の注意点
CellbenderのQCがうまく行かない場合はオプションをうまく調整していく必要があります。それぞれベストプラクティスに沿ったオプションが解説されているので参考にしてみてください。以下はドキュメントを日本語訳したものを載せておきます。
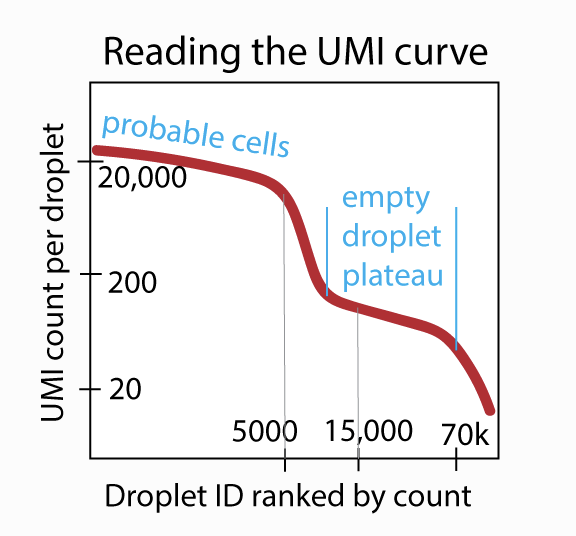
-epochs: 150が一般的に良い選択です。 出力されるPDFの学習曲線で、適度に収束したELBO値(ある飽和値に達したように見えるという意味)を探してください。より多くのエポック数を学習したくなるかもしれませんが、オーバーフィッティングの可能性が高くなるため、過剰な学習はお勧めできません。(オーバーフィッティングを防ぐために正則化を行いますが、300エポック以上の訓練はやりすぎです)。-expected-cells:実験デザインから事前に予想される細胞の数を基にするか、それがわからない場合は、以下に示すようにUMI曲線に基づいてこの数を設定します。UMI曲線上の左側の液滴がすべて実際の細胞であることが合理的に確認できる数を選ぶ。total-droplets-included:数千のバーコードが「空の液滴のプラトー」に入る数を選択します。 ただし、この数値が大きくなると、アルゴリズムの実行にかかる時間が長くなる(線形)ことに注意してください。 以下の UMI 曲線を参照し、適切な選択は 15,000 です。 UMI曲線でこの数値より右側の液滴はすべて確実に空になるはずです。(このようなUMI曲線はcellranger countのweb_summary.html出力で見ることができます。)cuda:このフラグを含めてください。 このコードはGPU上で実行されることを想定しています。-learning-rate:デフォルト値の1e-4は通常問題ありませんが、学習曲線の品質管理チェック(上記の通り)で問題が発生した場合は、この値を調整することができます。fprです:0.01の値は一般的にかなり良い値ですが、いくつかの値を選んで出力カウントマトリックスを生成し、比較することができます:0.01 0.05 0.1
最後に
いかがだったでしょうか。クオリティコントロールはscRNA-seqでは結果に大きく影響を与える大事なステップです。色々と試行錯誤してみてください。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます


{kind=link}