
RNA-seqを用いた全ゲノム解析のやり方を知りたいと思いませんか?RNA−seq解析はドライ系のバイオインフォマティシャンの人だけがやるんでしょ..?と考えている実験系の人は多いかもしれませんが、近年はRNA−seq解析の敷居が下がり、実験系の人でも取り入れることが可能になってきております。
でもRNA−seq解析ってPC上でどうやるんだろう?最近、公共データで全ゲノム解析ができると聞いたからやってみたいと感じている人向けに、今回は公共データとikraというツールを用いて、RNA−seq解析を自分のPCを用いてやる方法をご紹介したいと思います。
※本記事ではNGSを用いたRNA-seq実験のやり方は解説しません。あくまで、すでにシーケンス結果としてアーカイブされているものを使用します。
*2022年8月3日追記
コメント欄の方で、output.tsvはscaledTPMデータになっている点をご指摘いただきました。
参考:https://support.bioconductor.org/p/84883/
*2023年8月26日追記
本記事にはWindows版の解説記事がありますのでWindowsユーザーの方は参考にしてください。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,000 今なら60%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
ikraについて
ikraはRNA-seq解析を完全自動化したツール
ikraはRNA-seqの生データから、発現量データをTSVファイルとして出力する解析パイプラインです。RNA-seq解析を行うには複数のツールを組み合わせて段階的に解析していく必要がありますが、ikraではすべてのツールをパイプラインとしてつなぐことで、実行コマンド一つで自動的に各ツールが実行されていく仕組みになっています。そのため、RNA-seq解析初心者が各ツールの使い方がわからず挫折する、、といった事態を避ける事ができます。
またikraで使用できるRNA-seqデータの生物種はhumanかmouseに限られていますので、ご注意ください。
ikraから出力されるファイル群
ikraから出力されるファイルはクオリティチェックファイル(fast.qc)など複数ありますが、遺伝子発現の定量結果はoutput.tsvに出力されます。今回はこのoutput.tsvを出力するところまでを解説します。
ikraを使う準備
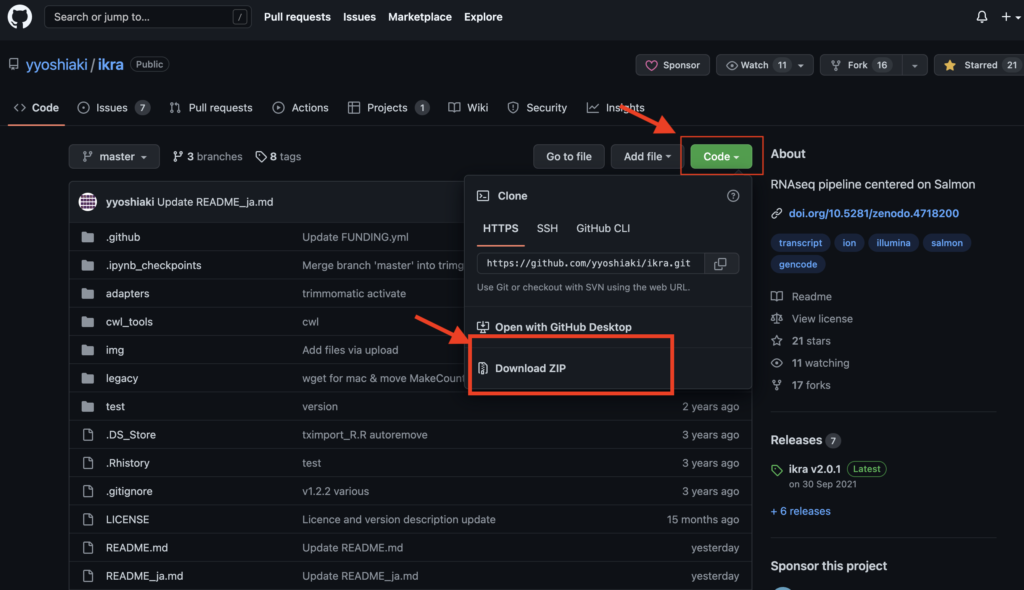
下記URLのGithubより「Code」→「Download ZIP」へ進みzipファイルをダウンロードし、zipファイルを解凍して中身を取り出します。zipファイルはデスクトップで解凍しておいてください。
- dockerのインストール
sra-toolkitのインストール
sra-toolkitsのインストールはMacとWindowsで手順が違いますので分けて記載します。
【Macの場合】
sra-toolkitのMacOSをインストールしてください。
インストールが終わったらsra-toolkitにPATHを通します。下記は3.0.0ですが、インストール時のバージョンに合わせて変更してください。
export PATH=$PATH:$PWD/sratoolkit.3.0.0-mac64/bin
which fasterq-dumpを叩いてみてsra-toolkitにPATHが通ってるか確認しましょう。

【Windowsの場合】
Ubuntuを起動します。デスクトップに移動しますが、自分の場合は以下のパスで移動できました。[ユーザー名]は適宜自身のものに適宜変更してください。
cd /mnt/c/Users/[ユーザー名]/desktop
sra-toolkitsをインストールしていきます。Ubuntu LTS 18.04以前の方は、updateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得してから、
sudo apt update -y
以下のコマンドでインストールすることができます。
sudo apt install sra-toolkit
Ubuntu LTS 18.04以降のバージョンの方は、古いファイルをインストールすることで使用できます。
sudo apt update -y
updateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得しておきます。
無事installができたらfasterq-dumpコマンドが通るか試してみましょう。以下のような画面になったら正しくインストールできています。

ここまでできたら、ikraを使う準備は完了です。
ikraへ読み込ませるデータ準備
解析したいSRAデータを探すの主に以下3つの場所があります。
- DBCLS https://sra.dbcls.jp/
- NCBI SRA https://www.ncbi.nlm.nih.gov/sra/
- Array Express https://www.ebi.ac.uk/arrayexpress/
今回はNCBI SRAを用いてデータを探索します。検索バーに探索したいキーワードを入力してください。
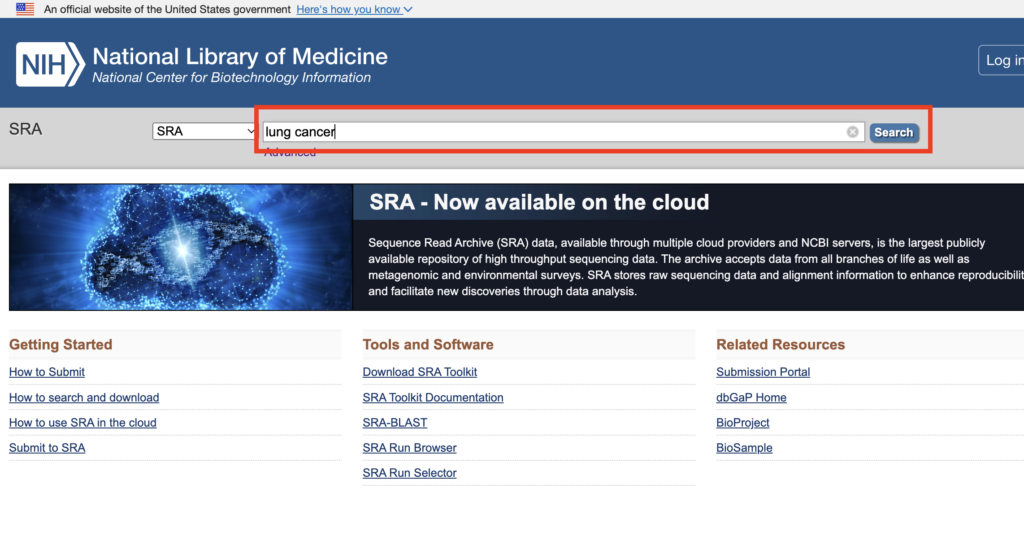
検索結果が出てきます。解析したいデータをクリックしてください。
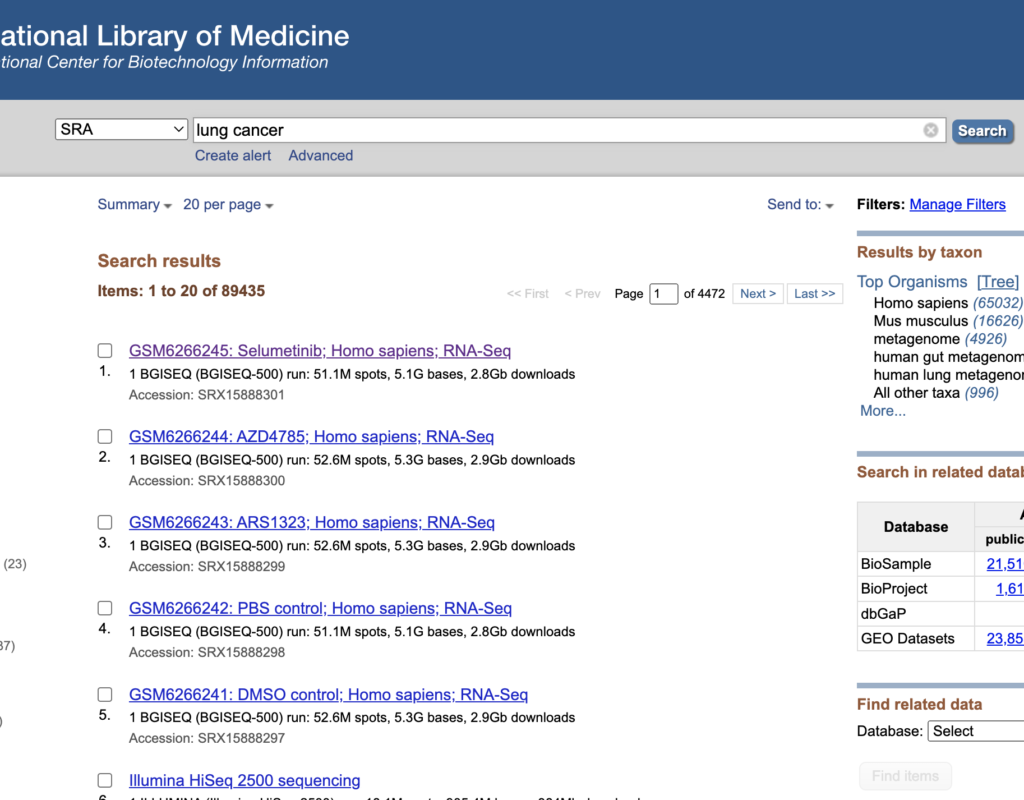
下にスクロールするとSRR~から始まる番号がありますので、こちらをクリックしてください。SRR番号は解析に用いるので、覚えておいてください。
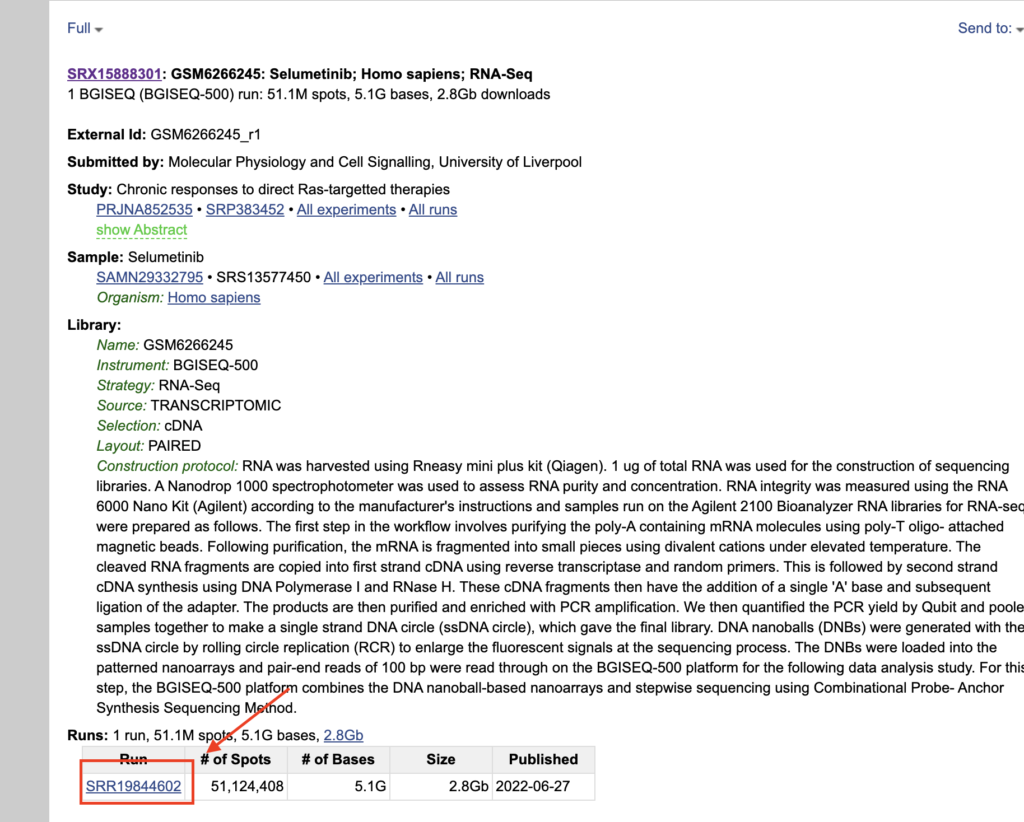
レイアウトがシングルエンドかペアエンドかを確認します。PAIREDになっていたらペアエンド、SINGLEになっていたらシングルエンドです。
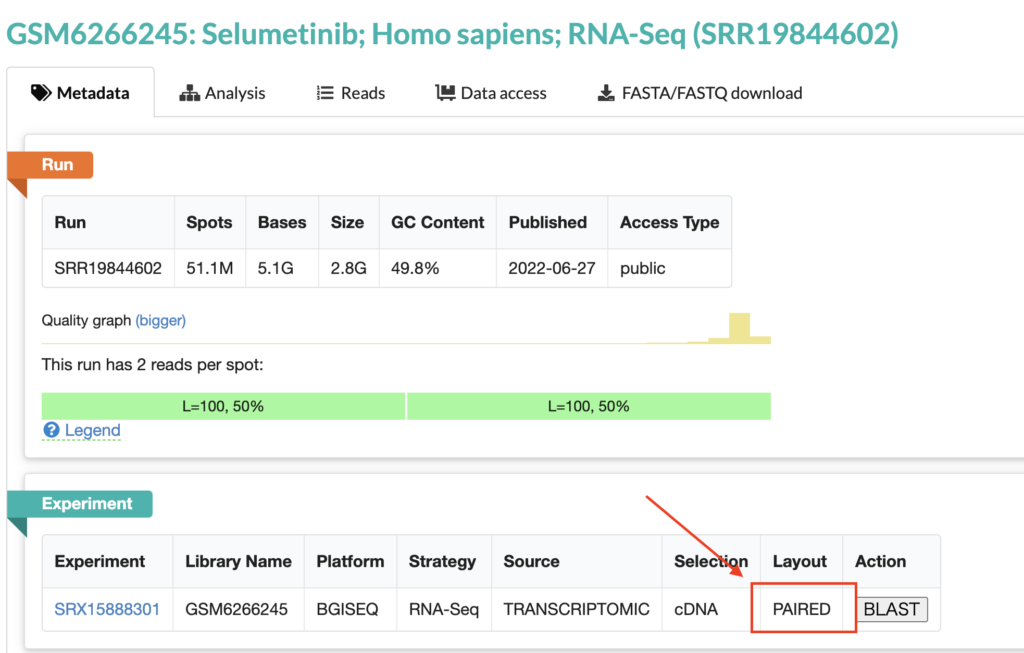
CSVファイルの作成
ikraに読み込ませるcsvファイルには以下の要素が必要です。name, SRR, Layoutという見出しを一行目に記載します。すべてコンマ(,)具切りで書きましょう。
- name・・好きな名前。何でも良い
- SRR・・SRR番号
- Layout・・ペアエンドかシングルエンドかの情報。ペアエンドは「PE」、シングルエンドは「SE」と表示します。
name,SRR,Layout
lung_cancer,SRR19844602,PECSVファイル用意ができたら、ikra.shファイルがあるディレクトリと同一のところに、csvファイルをおいてください。
実際にコードを実行してみよう
実際にikraを実行するときはコマンドライン(Macだとターミナル)で以下のコマンドを叩きます。
※コマンドを叩くときは、ikra.shがあるファイルのディレクトリに移動してから実行してください。(結果が出るまで4~10時間ぐらいかかります。)
bash ikra.sh 〇〇.csv mouse(or human)実行結果
output.tsvが出来上がっていると思いますので、見てみてください。遺伝子ごとのTPMデータが出来上がっていると思います。
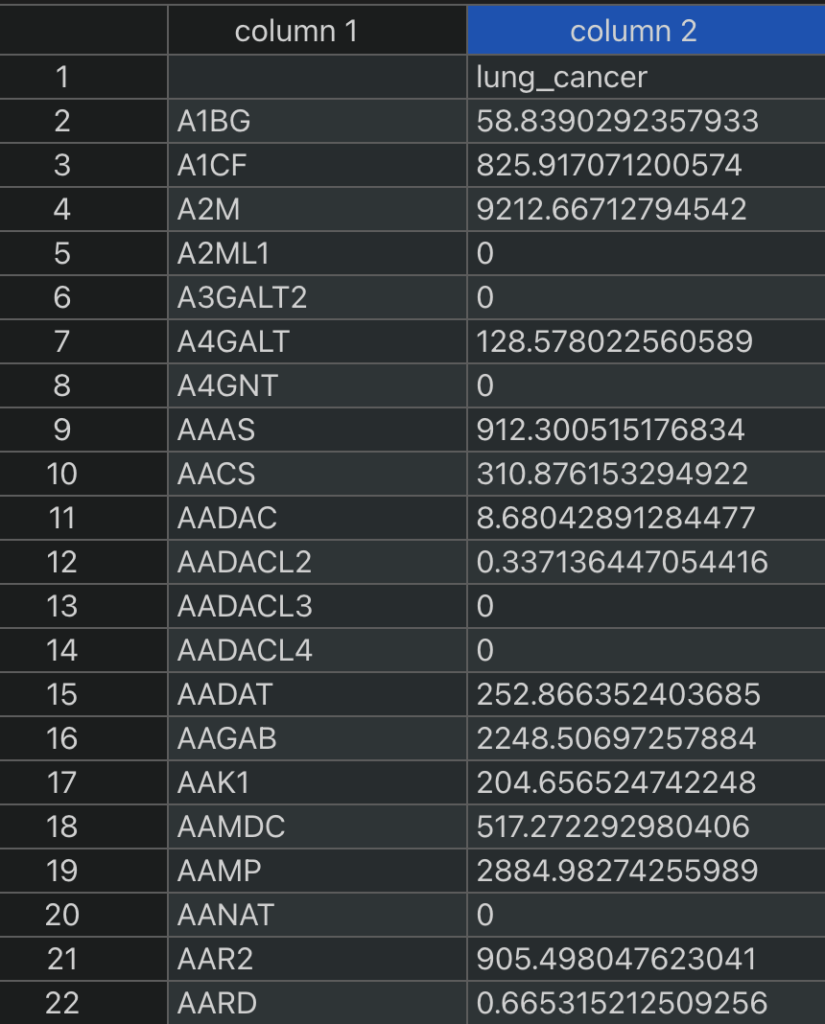
まとめ
いかがだったでしょうか。思ったより簡単にRNA−seqの定量結果を出力できたと感じた人もいるのでは無いでしょうか。公共データに登録されているRNA-seqデータは、コントロールのデータ群も登録されていますので、コントロール群との比較によって有意差検定も行うことが可能です。
TSVデータ以外にも、qcファイルなど他のファイルのデータの見方や、今回は数値データを出力するところまでやりましたが、出てきたデータを可視化する方法も別の記事で紹介したいと思います。
今回得られたTPMの数値データを、グラフィカルな図へと可視化する方法はいくつかあります。ikraで得られたRNA−seqデータをどのように可視化させていくかも次回ご紹介したいと思います。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,000 今なら60%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます


わかりやすくご紹介いただきどうもありがとうございます。ikraの作者です。
一点だけ、output.tsvはTPMではなくてscaledTPMである点にご注意ください。
DESeq2やiDEPの入力と使用する際、これらの入力値は負の二項分布を仮定しており、scaledTPMの使用が推奨されていたためです。
https://f1000research.com/articles/4-1521/v2
https://support.bioconductor.org/p/84883/
やすみず様
コメントいただきありがとうございます。記事執筆者です。ikra製作者であるやすみず様に本記事を読んでいただけて大変光栄です。
TPMではなくてscaledTPMである点のご指摘いただきありがとうございます。こちら、記事に注釈を入れて対応させていただきました。
記事に補足いただきたい点が他にもございましたら、お申し付けいただけましたら幸いです。
output.tsvを可視化して解析する方法も執筆予定でiDEP等取り上げようとしておりましたので、アドバイス頂いた点は是非参考にさせていただきます。