
Single Cell RNA-seqデータのQCには様々なパターンがありますが、その中の一つにダブレット検出があります。
本記事では、DoubletFinderというダブレット検出ツールを用いて、scRNA-seqデータからダブレットを検出してみようと思います。
ダブレットを検出できるようになれば、scRNA-seqデータ解析でより信頼性が増します。是非やってみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB, DoubletFinder 2.0.3

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
ダブレット検出とは?
ダブレットとは、2つまたはそれ以上の細胞が偶然一緒にキャプチャされ、単一の細胞として処理される現象を指します。これはシングルセルデータの解析において大きな問題となります。なぜなら、ダブレットは2つの異なる細胞タイプからの遺伝子発現パターンを混在させることで、細胞タイプの特定や遺伝子発現の解析を誤導する可能性があるからです。
10xgenomicsが公開しているQuality Controlのやり方を見てみると、コミュニティツールを使用したダブレット検出による細胞のフィルタリングについて、「ダブレットやマルチプレットは、解析を混乱させる可能性があります。DoubletFinder、Scrublet、Soloなどのツールがあります。」と述べられて言います。
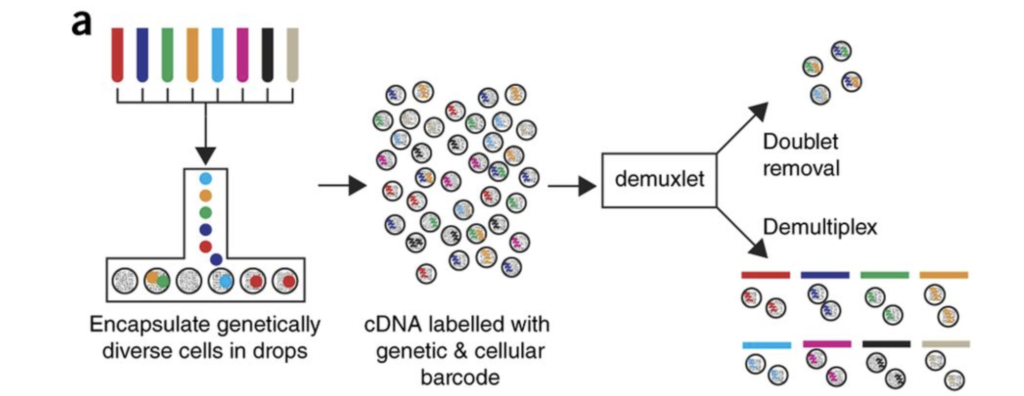
こちらの論文で、ダブレット検出の包括的なベンチマークを9つのツールで行った所、DoubletFinderは、最も高い検出精度と下流の解析における最大の改善効果を持つことが明らかにされています。
DoubletFinderは、以下のステップでダブレットを検出します:
- 人工的にダブレットを作り出す:データセット内の各細胞に対して、他の細胞からランダムに選ばれた遺伝子発現プロファイルを加えることで、人工的なダブレットを作成します。
- PCA(主成分分析)を実施し、ダブレットとシングルトン(一つの細胞のみからなるデータ)をプロットします:人工的なダブレットは通常、PCA空間においてシングルトンよりも遠くにプロットされます。
- 二項混合モデルを使用して、PCA空間におけるダブレットとシングルトンのクラスタを分離します。
- これらのクラスタを元に、各細胞がダブレットである確率を計算します。
以上のステップにより、DoubletFinderはシングルセルRNA-seqデータからダブレットを効率的に除去することができます。
DoubletFinderを使ってみる
では早速DoubletFinderを用いて、ダブレット検出を行ってみましょう。
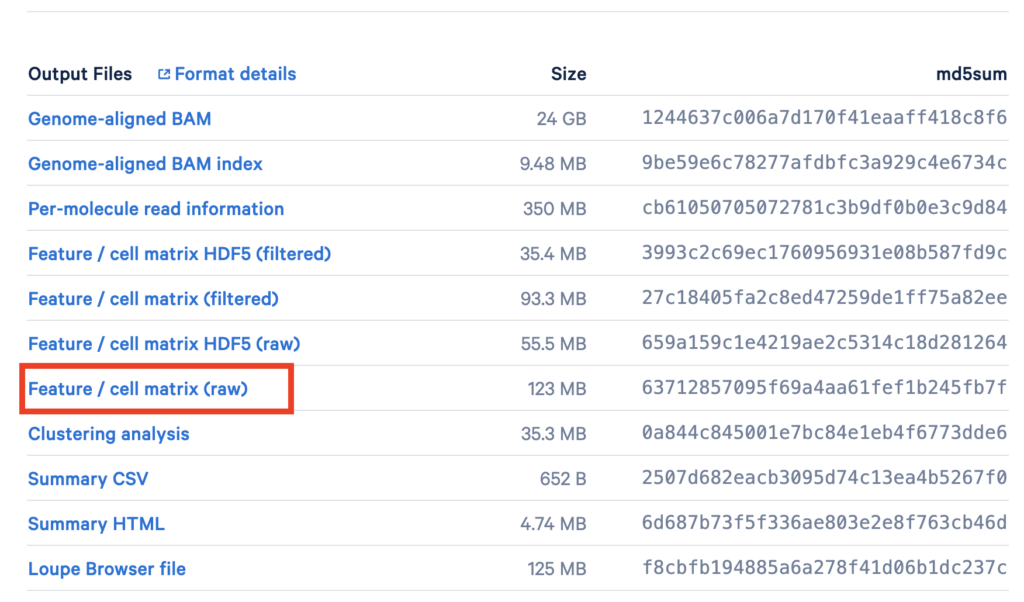
今回使うデータセットを10k Human PBMCs, 3′ v3.1, Chromium Controllerよりダウンロードしていきます。下にスクロールし、Feature / cell matrix(raw)をダウンロードしていきます。ダウンロードが終わりましたら解凍しておいてください。
必要なライブラリをインストールしていきます。
library(Seurat)
library(ggplot2)
library(tidyverse)
library(DoubletFinder)先程ダウンロードしたファイルを読み込んでいきます。UMAPまでの流れはいつも通りなので割愛させていただきます。操作の意味がわかっていない方は下記記事を参考にしてください。
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(前編)【Seurat】
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(後編)【Seurat】
# create counts matrix
cts <- Read10X(data.dir = "./raw_feature_bc_matrix/")
# create Seurat object
pbmc.seurat = CreateSeuratObject(counts = cts)
# QC and Filtering
# explore QC
pbmc.seurat$mitoPercent <- PercentageFeatureSet(pbmc.seurat, pattern = '^MT-')
pbmc.seurat.filtered <- subset(pbmc.seurat, subset = nCount_RNA > 800 &
nFeature_RNA > 500 &
mitoPercent < 10)
# pre-process standard workflow
pbmc.seurat.filtered <- NormalizeData(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- FindVariableFeatures(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- ScaleData(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- RunPCA(object = pbmc.seurat.filtered)
ElbowPlot(pbmc.seurat.filtered)
pbmc.seurat.filtered <- FindNeighbors(object = pbmc.seurat.filtered, dims = 1:20)
pbmc.seurat.filtered <- FindClusters(object = pbmc.seurat.filtered)
pbmc.seurat.filtered <- RunUMAP(object = pbmc.seurat.filtered, dims = 1:20)
doubletFinderでは、ダブレットスコアを計算するためにシミュレーションダブレットを作成します。これは、データセット内の異なるセル間で発現パターンが共有されている可能性のあるセルを模倣します。
シミュレーションダブレットを作成する際に使用するパラメータとして、pKとpN を計算します。
pKは、全体のセル集団に対するクラスタの割合を指定します。このパラメータは、各クラスタから選択されるシミュレーションダブレットの数を決定します。高いpK値を設定すると、より多くのシミュレーションダブレットが作成されます。pNは、シミュレーションダブレットを作成するために使用する期待されるダブレット率を指定します。たとえば、pN = 0.05は、元のシングルセルデータセットに5%のダブレットが存在すると予想されることを意味します。
下記のコードを実行することで、pK valueについて計算し、プロットを出せます。
find.pKを行うことで、最適なpKが計算されます。がここではpKが0.2のときにBCmetricが最大であることが分かりました。
## pK Identification (no ground-truth) ---------------------------------------------------------------------------------------
sweep.res.list_pbmc <- paramSweep_v3(pbmc.seurat.filtered, PCs = 1:20, sct = FALSE)
sweep.stats_pbmc <- summarizeSweep(sweep.res.list_pbmc, GT = FALSE)
bcmvn_pbmc <- find.pK(sweep.stats_pbmc)
ggplot(bcmvn_pbmc, aes(pK, BCmetric, group = 1)) +
geom_point() +
geom_line()生成したデータフレームbcmvn_pbmcから最適なpK値(最も高いダブレットスコア、BCmetric)を特定しています。ここでは先程のコードで計算された最適なpK値を格納しています。
pK <- bcmvn_pbmc %>% # select the pK that corresponds to max bcmvn to optimize doublet detection
filter(BCmetric == max(BCmetric)) %>%
select(pK)
pK <- as.numeric(as.character(pK[[1]]))このコードはdoubletfinderパッケージの機能を用いて、ダブレット(同じウェルに二つ以上の細胞が存在する状態)を検出するための準備を行っています。
## Homotypic Doublet Proportion Estimate -------------------------------------------------------------------------------------
annotations <- pbmc.seurat.filtered@meta.data$seurat_clusters
homotypic.prop <- modelHomotypic(annotations) ## ex: annotations <- seu_kidney@meta.data$ClusteringResults
nExp_poi <- round(0.076*nrow(pbmc.seurat.filtered@meta.data)) ## Assuming 7.5% doublet formation rate - tailor for your dataset
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
annotations <- pbmc.seurat.filtered@meta.data$seurat_clustersでは、Seuratオブジェクト(ここではpbmc.seurat.filtered)のメタデータからクラスタリングの結果(seurat_clusters)を取得し、annotationsに保存しています。これにより、各細胞がどのクラスタに所属しているかの情報を取得しています。homotypic.prop <- modelHomotypic(annotations)では、doubletfinderパッケージのmodelHomotypic**関数を使用して、同一クラスタ内(homotypic)のダブレットが存在する割合を推定しています。nExp_poi <- round(0.076*nrow(pbmc.seurat.filtered@meta.data))では、全細胞数に対するダブレットの割合(ここでは7.6%と仮定)を用いて、期待されるダブレット数を計算しています。nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))**では、同一クラスタ内のダブレットの存在割合を考慮して、期待されるダブレット数を調整しています。この値はダブレット検出アルゴリズムで使用されます。
いよいよ下記コードでDoublet Finderを実行します!
# run doubletFinder
pbmc.seurat.filtered <- doubletFinder_v3(pbmc.seurat.filtered,
PCs = 1:20,
pN = 0.25,
pK = pK,
nExp = nExp_poi.adj,
reuse.pANN = FALSE, sct = FALSE)
実行した結果をDimplotで可視化してみましょう。
# visualize doublets
DimPlot(pbmc.seurat.filtered, reduction = 'umap', group.by = "DF.classifications_0.25_0.22_691")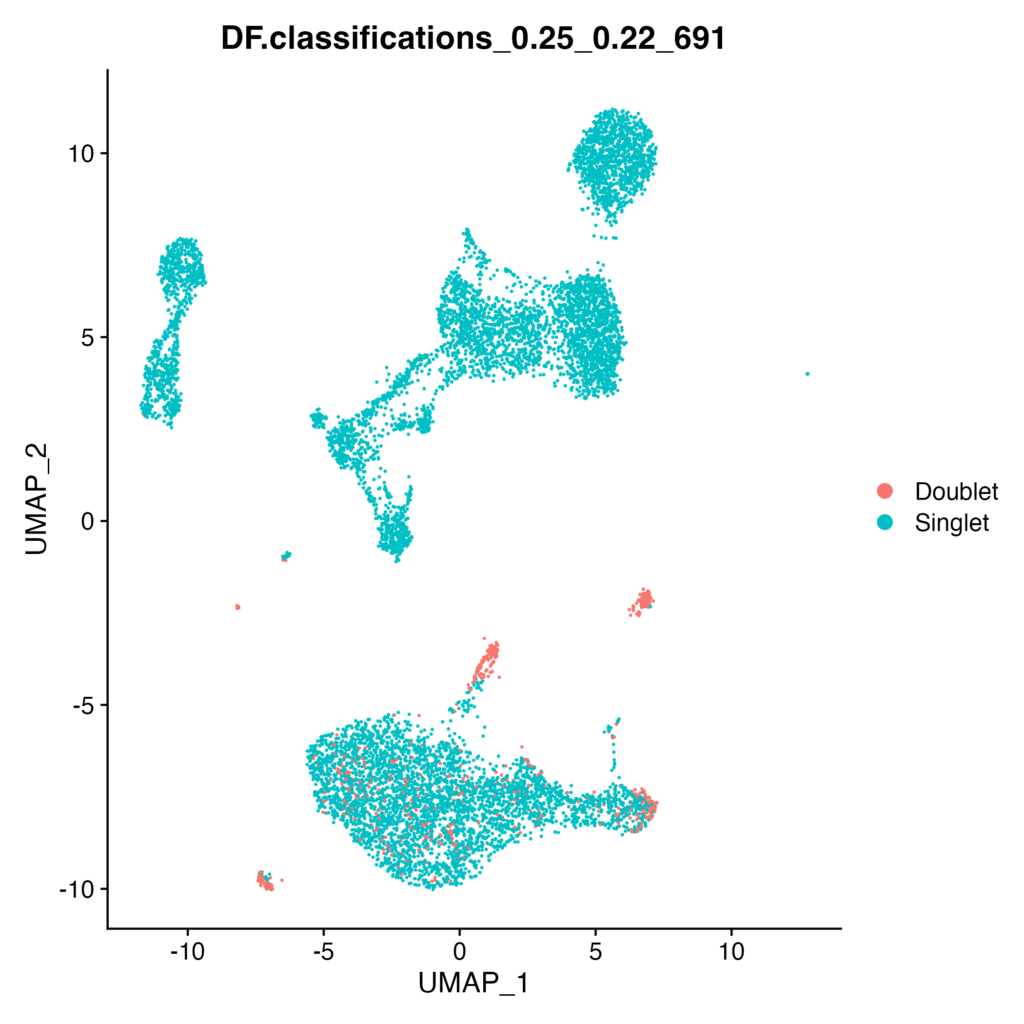
Doublet として検出されたものが赤色で示されています。
最後にDoubletの数を出力してみましょう。
# number of singlets and doublets
table(pbmc.seurat.filtered@meta.data$DF.classifications_0.25_0.22_691)
### 出力
Doublet Singlet
691 9326 Doubletが691含まれていることが分かりました。
最後に
いかがだったでしょうか。DoubletFinderをつかってscRNA-seqデータ解析をもっと信頼性のおける解析にしていきましょう。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

