
This blog post is about in silico screening of small molecules in the field of drug discovery. It is used to identify candidate compounds for target molecules. By learning the content of this article, you can experience a series of in silico drug discovery processes, including protein preparation, small molecule library preparation, and in silico screening. Please give it a try!
windows 64 bit, PyMOL 2.5.4. PyRx 0.8
What is In Silico Screening?
In silico screening is a type of virtual screening that uses computer simulations to select promising compounds from large molecular libraries for specific biological targets.
In silico screening, computer models are used to predict the structural information and biological activity of a large number of compounds, enabling the search for optimal compounds against the target of interest.
This process allows the selection of promising compounds before experimental synthesis, saving effort and cost. It is also faster than experimental screening and enables the simultaneous evaluation of a larger number of compounds, thereby enhancing the efficiency of drug development.
In this case, due to the limitations of computer specifications, we will perform in silico screening using a small library consisting of three compounds.
What is PyRx?
PyRx is a free molecular docking software that provides tools for virtual screening.
PyRx provides a graphical user interface (GUI) for easy preparation, execution, and analysis of molecular docking. It allows the use of standard formats for molecular structure data, making it easy to import compounds from various databases.
PyRx is available for free and supports multiple operating systems such as Windows, macOS, and Linux. This makes it a widely used virtual screening tool.
To proceed with this article, please download PyMOL.
PyRx Installation
First, download PyRx-0.8-Setup.exe from the PyRx download page for Windows. Note that there is also a Mac version, but it is currently not supported.
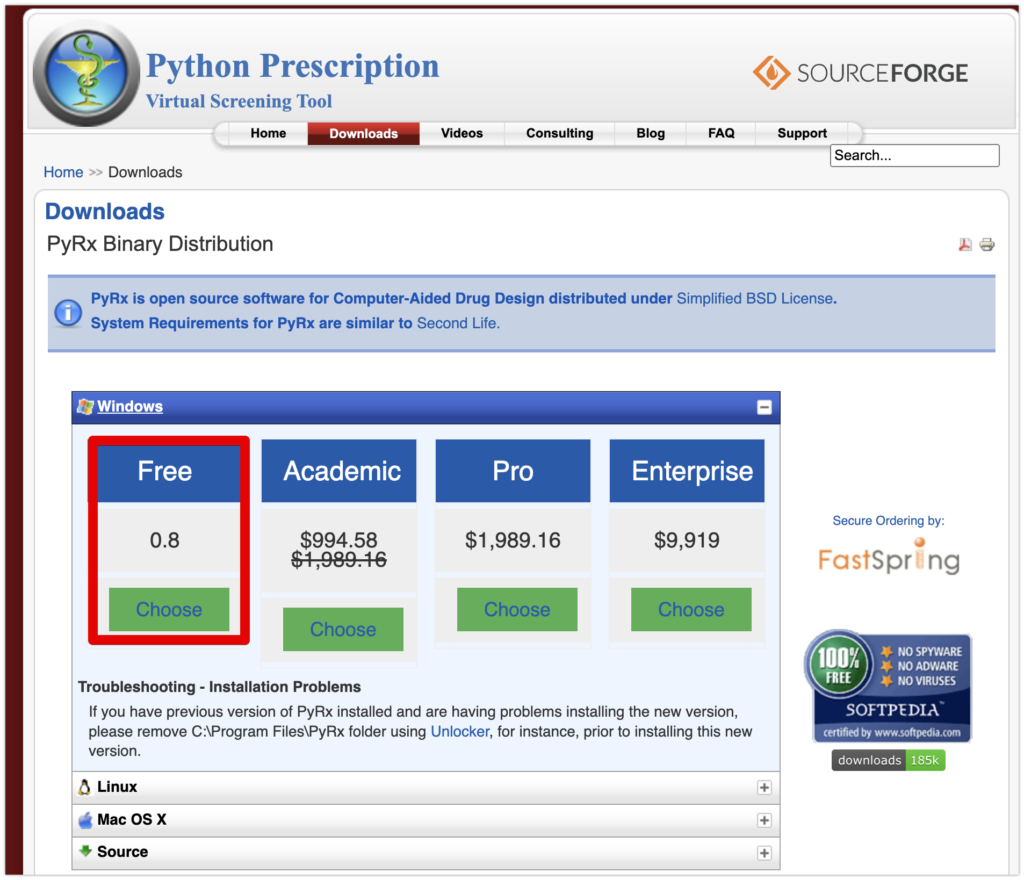
After downloading, install PyRx by running PyRx-0.8-Setup.exe. Simply click “Next” without making any special changes during the installation.
Protein Preparation
Next, we will prepare the target for in silico screening. Many proteins in the Protein Data Bank are complexes with binders, so we need to remove them. Here, we will use DPP4 (PDB: 2OQV), a target for diabetes, as an example.
Open PyMOL,
Click “File” -> “GetPDB…” and enter 2OQV as the PDB ID, then click “Download” to display DPP4 in PyMOL.
Next, since DPP4 consists of two identical units of proteins, we will select only one unit.
In the PyMOL command line, enter:
sele chain A
and execute it. “sele chain A” is the command to select molecules with chain ID A.
Also, execute:
color red, sele
to make chain A appear in red for clarity.
Since chain A contains the binder, we will remove it.
From “Display” -> “Sequence,” display the sequence and select MA9 on the far right.
Remove the binder by selecting “sele” -> “remove atoms.”
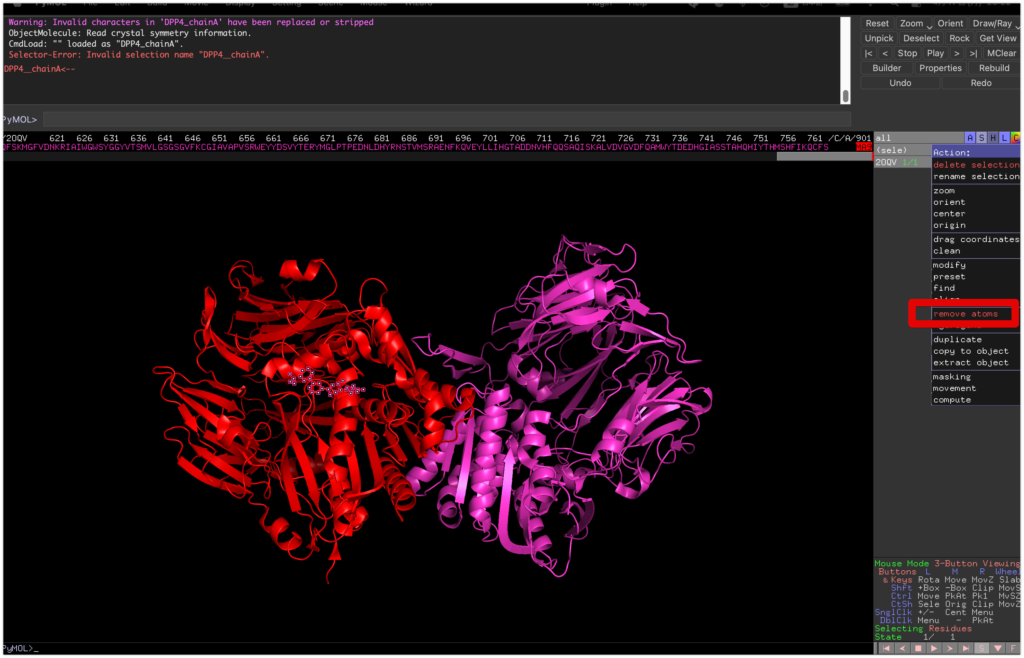
Then, select chain A again from “sele chain A,”
save DPP4_prep.pdb, sele
and save it as “DPP4_prep.pdb, sele” to save chain A as a PDB file named DPP4_prep.pdb.
This completes the protein preparation.
Preparation of Small Molecule Library
Next, we will prepare the small molecule library for screening. The process involves:
- Obtaining compound data in SDF format from PubChem or the ZINC database.
- Converting the structure data to PDB format.
1. Obtaining compound data in SDF format from PubChem or the ZINC database
PubChem is an open chemical database managed by the National Center for Biotechnology Information (NCBI), a division of the National Institutes of Health (NIH) in the United States. It provides comprehensive resources of chemical data, including biological activity, properties, and structures of millions of chemical compounds.
PubChem contains a wide range of chemical data, including chemical structures, properties, biological activities, safety data, and references to scientific literature. It is used for various purposes in the fields of chemistry, biochemistry, pharmacology, toxicology, and more. Examples include drug discovery, chemical research, and toxicity evaluation.
PubChem offers multiple search options based on chemical names, structures, properties, and biological activities. It also provides tools for analysis, visualization, and downloading of chemical and biological data.
First, go to PubChem and retrieve the desired small molecule compounds for the library.
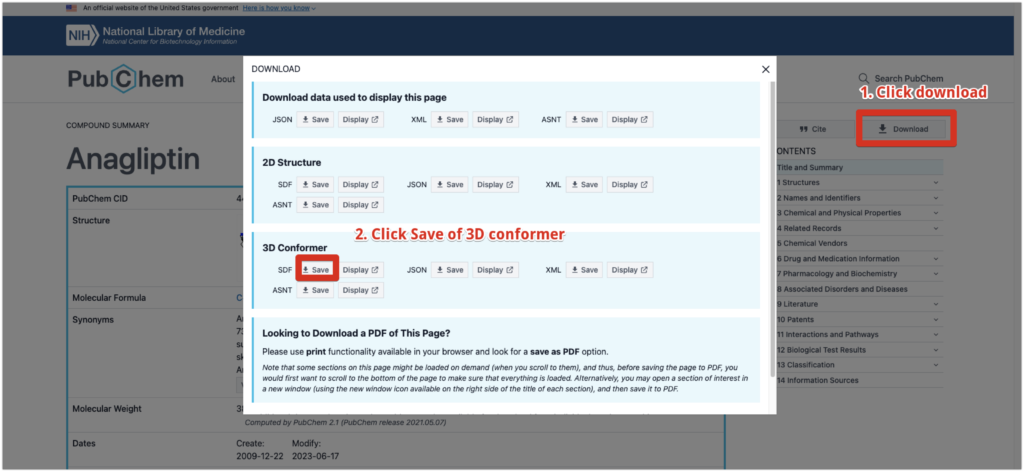
Here, we will use Anagliptin, a compound known to have an effect on DPP4, as an example.
Go to the page for Anagliptin and click “Download” -> “3D conformer” on the right to obtain the SDF file.
Alternatively, you can use the ZINC database to retrieve compound information, similar to PubChem.
The ZINC database is one of the publicly available chemical databases used for virtual screening of chemical compounds. It provides data and information on chemical compounds, assisting in drug discovery and design.
The ZINC database is one of the publicly available chemical databases used for virtual screening of chemical compounds. It provides data and information on chemical compounds, assisting in drug discovery and design.
The ZINC database is primarily used in the technique called virtual screening. Virtual screening is a method that involves rapidly screening a large number of chemical compounds on a computer to select promising compounds. The ZINC database is widely used for such virtual screening, supporting the search and design of new drugs as a useful tool.
Access the ZINC website, click on the “Substances” tab.
From here, you can retrieve the desired small molecule compounds.
Here, we will use Omarigliptin, a compound known to have an effect on DPP4, as an example.
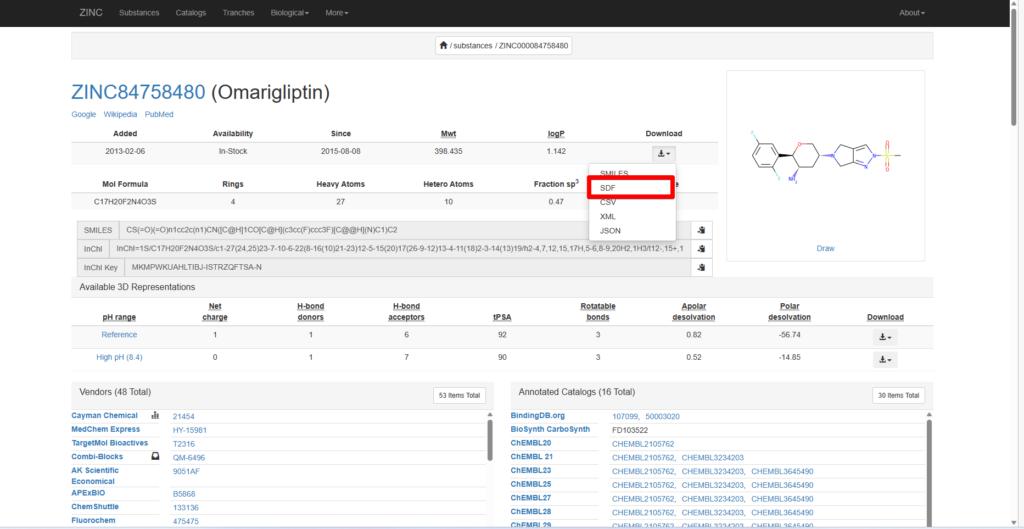
Search for Omarigliptin in the search bar, select Omarigliptin, and the following page will appear.
Click “Download” and save the SDF file.
2. Converting the structure data to PDB format
Next, we will convert the SDF file to PDB format. Open the downloaded file in PyMOL.
Click “File” -> “Export Molecule…” and the following screen will appear.

Click “Save” as it is, select PDB (*.pdb, *.pdb.gz) as the file format, and save the data as a PDB file.
Repeat this process for all compounds in the library.
In silico screening
Due to computer specifications, we will screen using three compounds: Anagliptin, Omarigliptin, Voglibose(an α-glucosidase inhibitor targeting a different aspect of diabetes).
First, go to the Program Files (×86) folder and select PyRx of VBScript files to launch it.
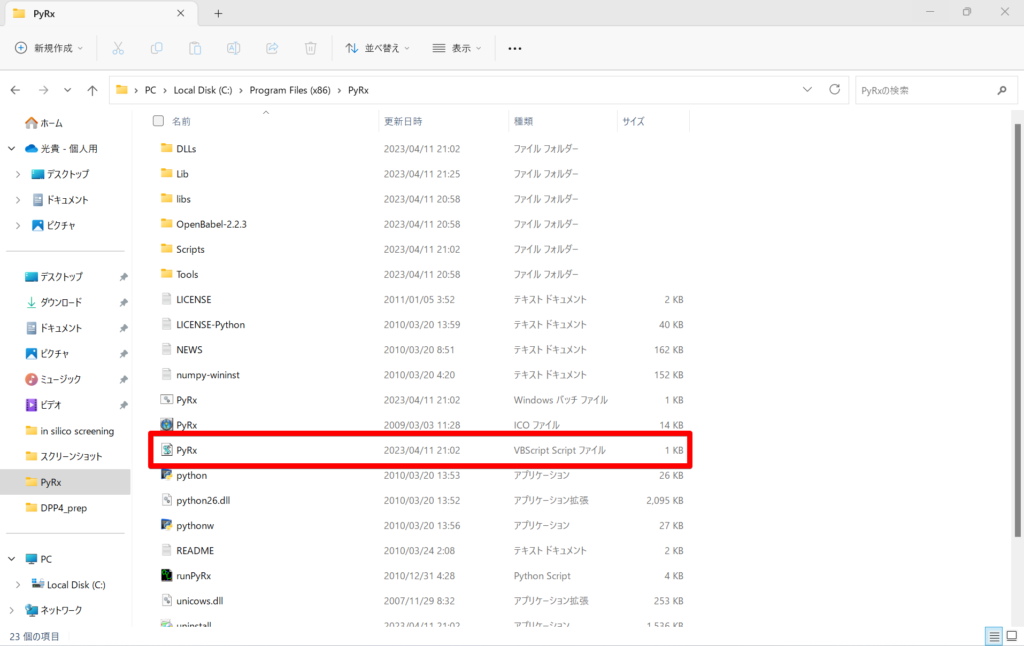
When it starts, you will see the following screen.
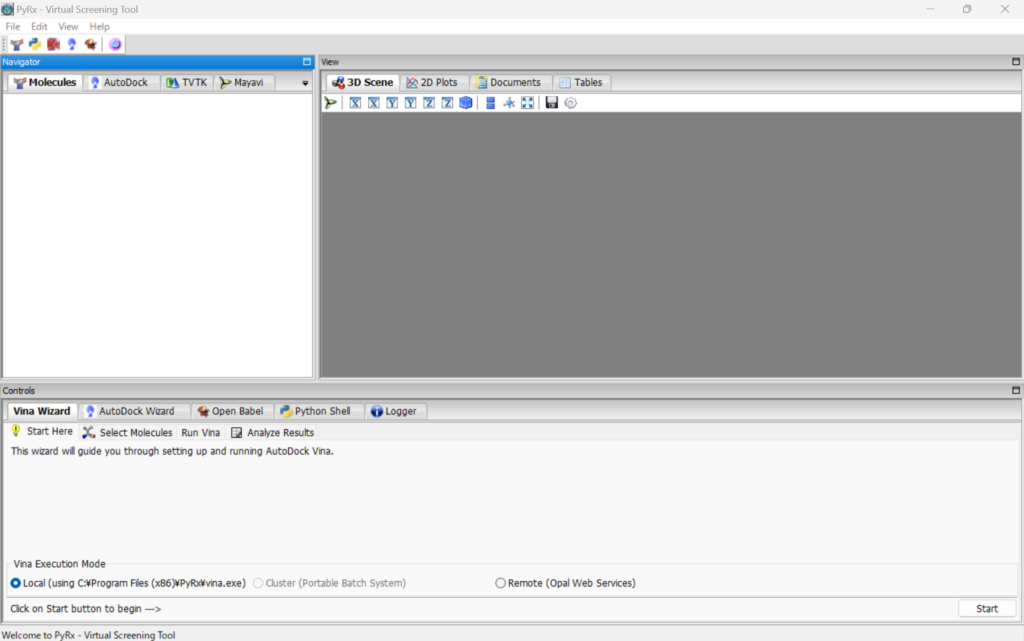
Right-click on the “Molecules” panel, select “Load Molecule,” and load all the protein and small molecule PDB files.
Next, right-click on DPP4_prep, select “AutoDock” -> “Make Macromolecule.”
Then, right-click on each small molecule and select “AutoDock” -> “Make ligand.”
Looking at the AutoDock toolbar, you can see that they are placed in “Macromolecules” and “ligands.”
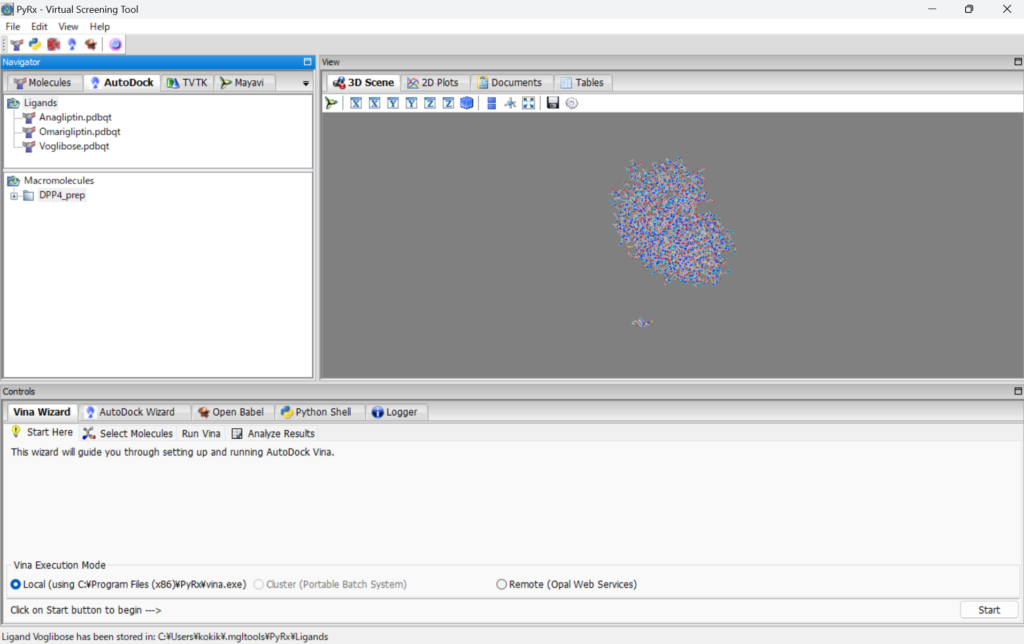
Then, click “start” in the lower right corner, select the small molecules and the protein. You will see the following display.
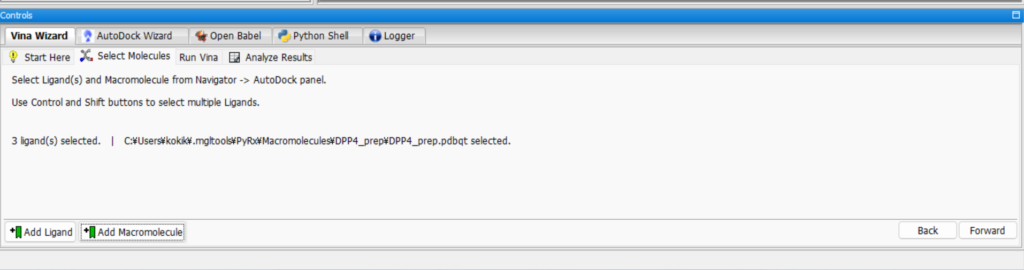
Leave it as it is, click “Forward,” adjust the 3D Scene rectangle, and select the binding site on the small molecule. By making sure the protein fits inside the rectangle, you can find the binding site where the small molecule can bind from various locations on the protein.
Then, click “Forward,” and in silico screening will start.
You will see a screen like the one below, so please wait for about 10 minutes.
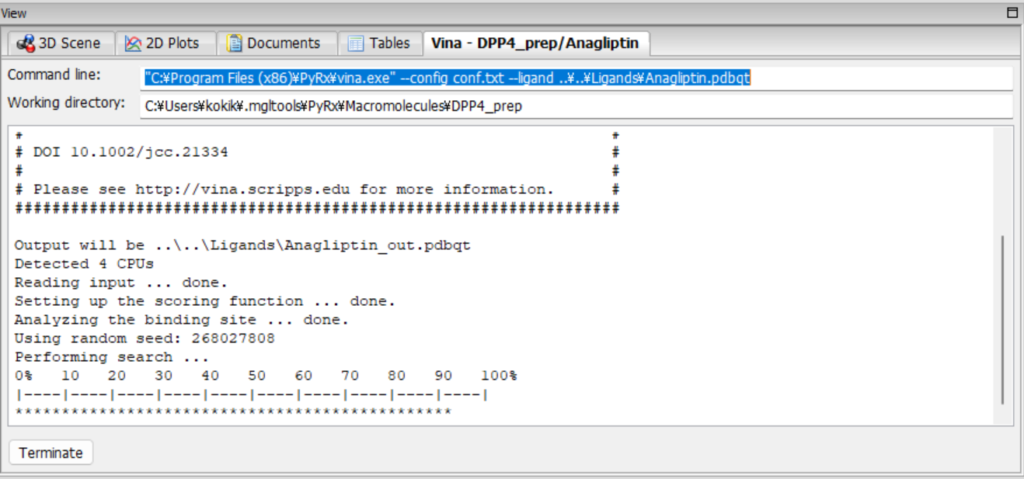
After a while, the results will appear. The following shows the results for DPP4 and Anagliptin.
Among these, the top result with the lowest “Binding Affinity (kcal/mol)” indicates the strongest binding.
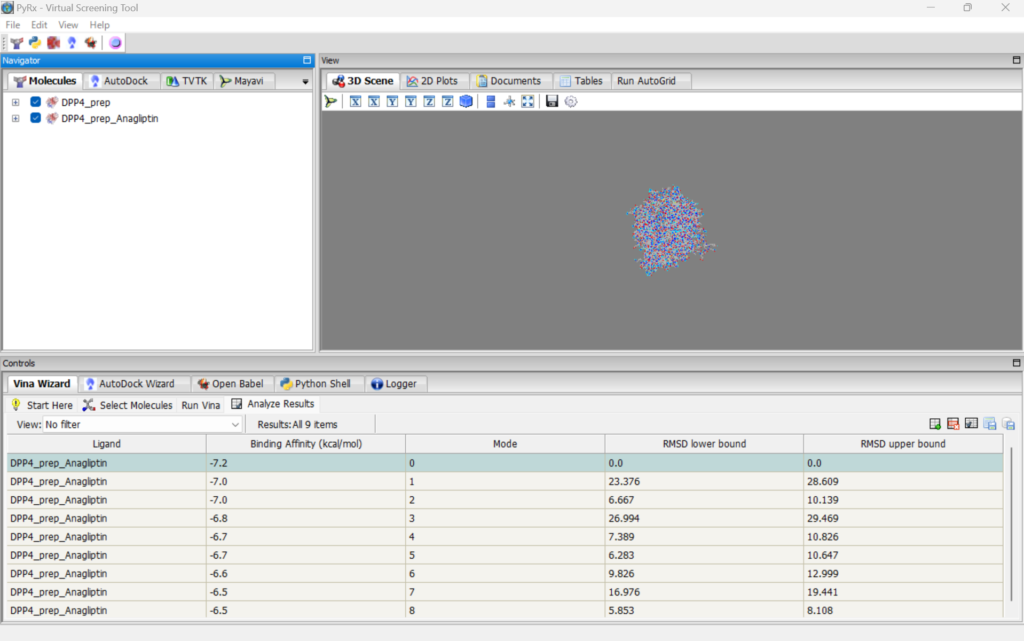
Additionally, among the compounds in the library, the lowest “Binding Affinity (kcal/mol)” values were: Anagliptin: -7.2, Omarigliptin: -7.6, Voglibose: -5.4 kcal/mol. This result indicates that Omarigliptin and Anagliptin, which are already known to have effects, exhibit higher binding affinity, confirming their strong binding capabilities.
Visualization of Binding Sites Using PyMOL
Finally, let’s use PyMOL to visualize where the screened compounds bind.
Check the workspace in “Edit” -> “Preferences” in PyRx to see if the results are outputted.
Go to C:\Users\kokik.mgltools\PyRx and navigate to Macromolecules -> DPP4_prep. You will find DPP4_prep, Omarigliptin_out, Anagliptin_out, and Voglibose_out. These represent the compounds that show the highest binding affinity at specific locations. Select them from PyMOL’s “File” -> “Open” to display them.
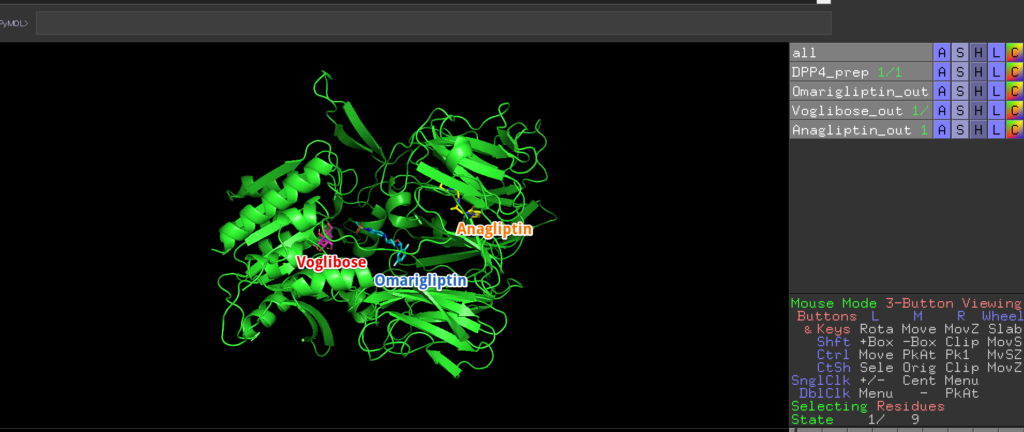
You can now see the binding sites for each compound! They all reside within DPP4, and it’s very intriguing!
Final Remarks
How did it go? In this article, we introduced one of the drug discovery methods, in silico screening. Try applying it to your favorite proteins and libraries to search for potential drug compounds!
Furthermore, although we did not utilize it this time, one advantage of the ZINC database is that you can search existing compound libraries from the “Catalog” -> “subsets” in the above toolbar. This allows you to easily search for compounds approved by the FDA.
catalog/subsets page
With this, you can perform large-scale screening with a variety of compounds. If you have a high-performance computer, give it a try!
Reference Videos
Protein-ligand virtual docking using PyRx | Computational biology | Bioinformatics | Akash Mitra
PyRx Tutorial || Multiple Ligand Docking || From Download to Result Analysis || All in One
PyRx – Virtual Screening Tool | Multiple Ligand Docking | Lecture 42 | Dr. Muhammad Naveed