
single cell RNA-seq解析(scRNA-seq)とはRNA-seq解析を個々の細胞集団ごとに行う手法として注目を集めています。
今回はscRNA-seqのRパッケージであるSeuratのハンズオンに取り組むことで、scRNA-seqを始める前準備を行おうと思います。
本記事はSeuratのハンズオンに則って行いますので、Seuratの使い方の概要を理解することが可能です。ぜひ取り組んでみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
Seuratとは?
SeuratはscRNA-seqデータ解析を行うRパッケージのことで、scRNA-seqデータの正規化やビジュアライズなどを行う事ができます。
Seuratには製作者たちによってハンズオンが用意されています。こちらに取り組むことでSeuratの理解を深めることができますが、Rに詳しくない方が取り組むには難しいところがあるため、こちらの記事では補足説明を加えつつ解説していきたいと思います。
R環境の用意
Seuratのハンズオンを行うにはRを実行する環境が必要となります。私的には、Rを実行する環境としてRstudioを使うことをおすすめします。
各OS環境を元にこちらのサイトよりインストーラーをダウンロードしてください。
https://cran.r-project.org/
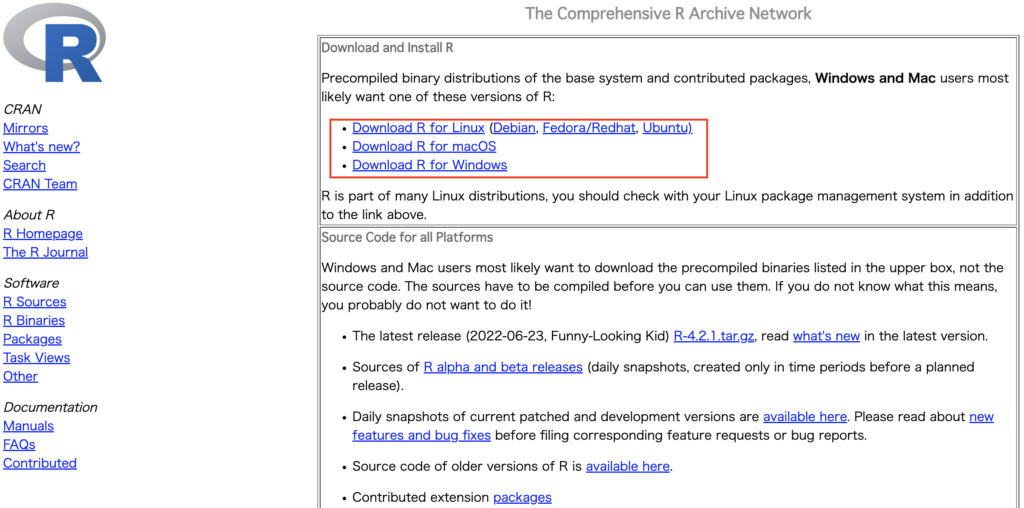
インストールするバージョンは最新版を選んでください。一番トップにあるものが最新版です。

インストール画面では「続ける」を押して完了させてください。
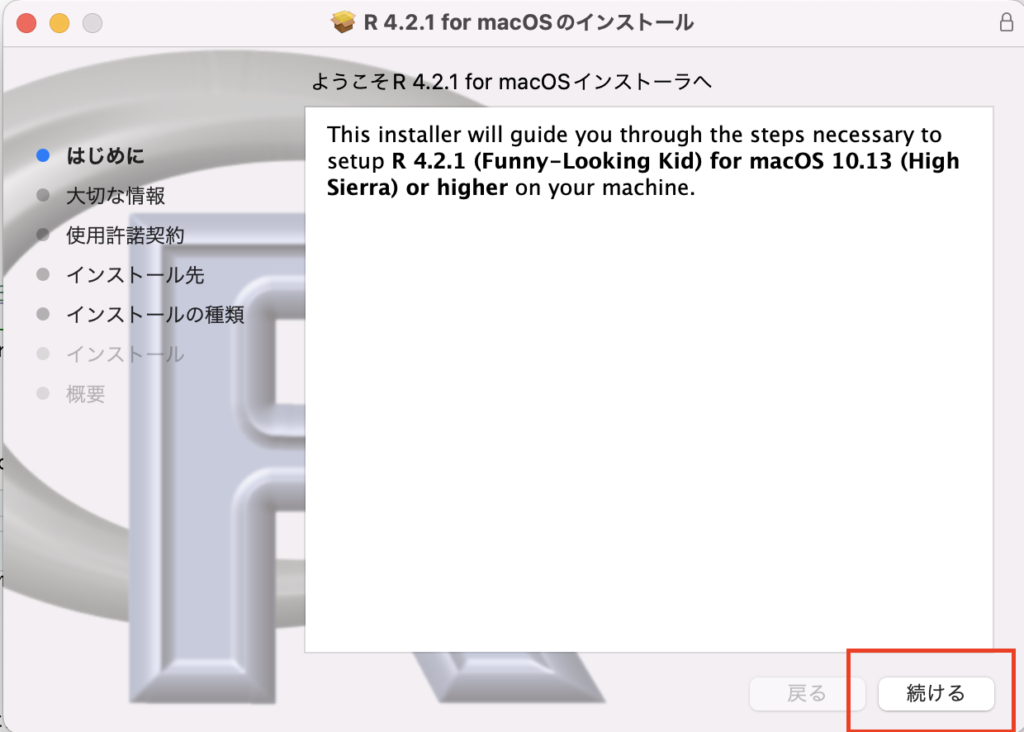
これでRのインストールは完了です。
Rstudioのインストール
こちらのURLにアクセスし、下に少しスクロールすると、「DOWNLOAD RSTUDIO」ボタンがあるのでクリックします。

以下のページに飛ぶと思います。2: Install RStudioのボタンをクリックします。画像はMacですが、このページに飛んだ時点で、WindowsならWindows用のRstudio Desktopがダウンロードできるようになっています。ダウンロードができたら、Rのときと同様にインストールを完了させてください。

Rstudioを開くと以下のようになっていると思います。

RStudioのインターフェイスは4つの主要なエリアに分かれています。
- ソースエディタ (左上のエリア)ここではRのスクリプトやR Markdownファイルを作成、編集することができます。また、Rのコードをここに記述し、「Run」ボタンをクリックするか、ショートカットキー(WindowsではCtrl+Enter、MacではCmd+Enter)を使用して実行することができます。
- コンソール (左下のエリア)ここではRコードを直接実行したり、実行結果を確認したりすることができます。また、Rのコマンドを試したり、関数のヘルプを見たりすることも可能です。
- 環境/履歴 (右上のエリア)
- 環境タブ: ここでは作業スペース内のオブジェクト(変数、データフレーム、リストなど)を表示します。新しいオブジェクトが作成されると、ここにリストされます。
- 履歴タブ: これまでに実行したRのコマンドの履歴を表示します。過去のコマンドを再利用するためには非常に便利な機能です。
- ファイル/プロット/パッケージ/ヘルプ (右下のエリア)
- ファイルタブ: ワーキングディレクトリ内のファイルを表示します。
- プロットタブ: グラフィカルな出力(プロットやチャート)を表示します。
- パッケージタブ: インストールされているRのパッケージを表示します。ここからパッケージをロードしたり、新たにパッケージをインストールしたりすることができます。
- ヘルプタブ: Rの関数やパッケージのヘルプドキュメントを表示します。これは、特定の関数の使用方法を確認したいときに役立ちます。
これらのエリアはそれぞれ独立して機能し、互いに連携してRのコーディングをサポートします。RStudioのレイアウトはカスタマイズ可能で、これらのパネルを移動したり、サイズを変更したりすることができます。
Rstudioではソースエディタ (左上のエリア)にコードを書いていきます。Runを押すと行実行ができます。Sourceを押すとスクリプト全体が実行できますので覚えておいてください。

Seuratの使い方
ハンズオンの準備
それではSeuratのハンズオンをやっていきたいと思います。まずは必要なライブラリをインポートしてください。
install.packages("dplyr")
install.packages("Seurat")
install.packages("patchwork")インストールが終わったら、以下の記述を記載してライブラリが使える状態にします。
library(dplyr)
library(Seurat)
library(patchwork)生データをハンズオンサイトからダウンロードして来ます。
こちらにアクセスし、図のhereからダウンロードしてください。
https://satijalab.org/seurat/archive/v3.2/pbmc3k_tutorial.html#:~:text=data%20can%20be%20found%20here
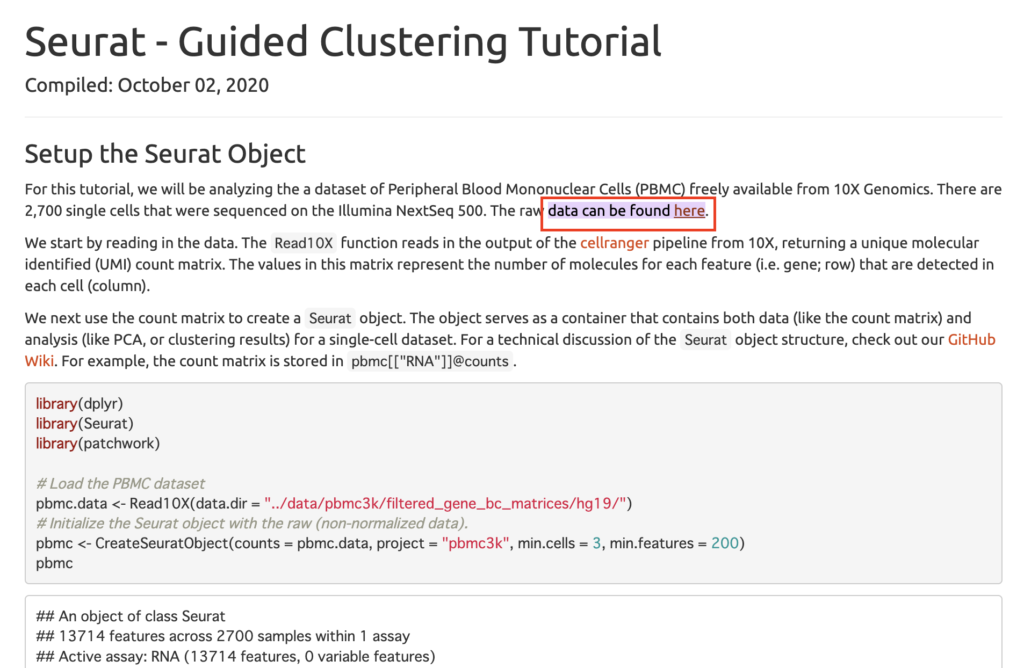
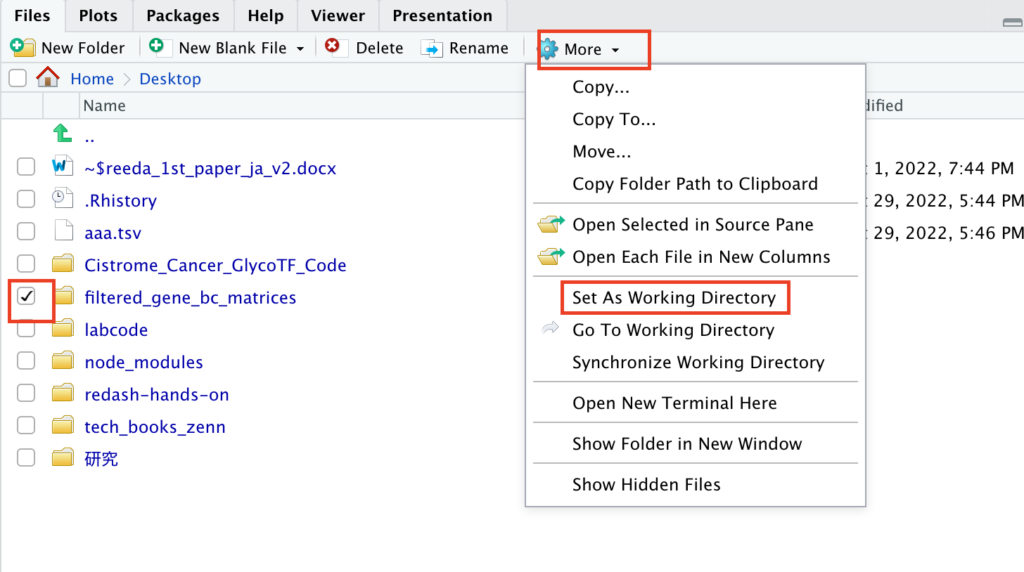
ダウンロードしてきたファイルをデスクトップ上で解凍してください。その後Rstudioの右下画面でデスクトップまで進み、
- ダウンロードしてきたファイルにチェック
- Moreをクリック
- Set As Working Directory
を順に選びます。この作業を間違うと後のコードが一切通らなくなるのでお気をつけください。
以下のコマンドを叩いて最後にDesktopで終わるようなパスが出てきたらきちんと設定がなされています。
> getwd()
[1] "/Users/[ユーザー名]/Desktop"以下のコマンドを実行し、データをRに読み込んでいきます。これでハンズオンの準備は完了です。
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmcクオリティコントロール(QC)
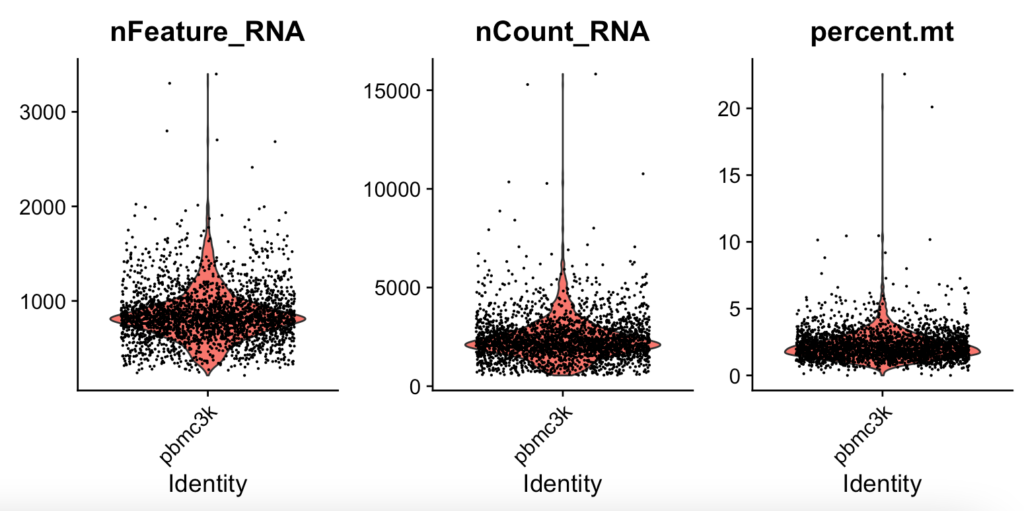
scRNA-seqではバイオリンプロットを用いてクオリティコントロールを行い、外れ値に存在する細胞がいるかを確認していきます。
細胞が死んでいるなど低いクオリイティのセルは遺伝子発現量が極端に低かったりミトコンドリア由来のコンタミが懸念されます。また遺伝子数カウントする際に細胞がダブる(シングルセルではない)と異常な遺伝子数でカウントされます。
# MT-で始まる全ての遺伝子の集合をミトコンドリア遺伝子の集合として使用する
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)こちらを打ち込むと以下のような図がRstudioの右下に現れると思います。
バイオリンプロットの膨らみが確認できる場所の細胞群を用いると全体としてもっともらしい遺伝子数カウントがされると判断します。今回のサンプルでのQCの参考値はいかになります。
nFeature_RNAより、ユニークフィーチャー数が2,500以上、または200以下の細胞をフィルタリングします。percent.mtよりミトコンドリア数が5%以上の細胞をフィルタリングします。
それぞれの相関関係を見るために散布図を利用することができます。下記のコードを実行してみてください。
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2percent.mtとnCount_RNAでは相関が見られません(-0.13)が、nFeature_RNAとnCount_RNAとは相関関係がある(0.95)ことがわかるかと思います。

QC後に決まった値を再度サンプルpbmcに入れておきます。
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)データの正規化
QCによってデータセットから不要な細胞を除去した後は、データの正規化を行います。
デフォルトでは、各細胞の特徴的な発現量を総発現量で正規化し、これにスケールファクター(デフォルトでは10,000)を掛け、結果を対数変換するグローバルスケーリング正規化手法 “LogNormalize” が採用されています。
データの正規化後に決まった値を再度サンプルpbmcに入れておきます。
# LogNormalize
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# pbmcに再度入れる
pbmc <- NormalizeData(pbmc)ここまでできれば、scRNA-seqデータの前処理は完了になります。後の処理は正規化されたデータの可視化方法の解説になります。
変動量の大きい遺伝子を抽出
ある細胞では高発現、別の細胞では低発現しているデータセット中の細胞間変動が大きいサブセットを計算します。デフォルトでは、1データセットあたり2,000のVariable countをを返すようになっています。
# FindVariableFeatures関数を使ってデータを用意
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# top10の遺伝子名を記述のため、headを取得
top10 <- head(VariableFeatures(pbmc), 10)
# プロット
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2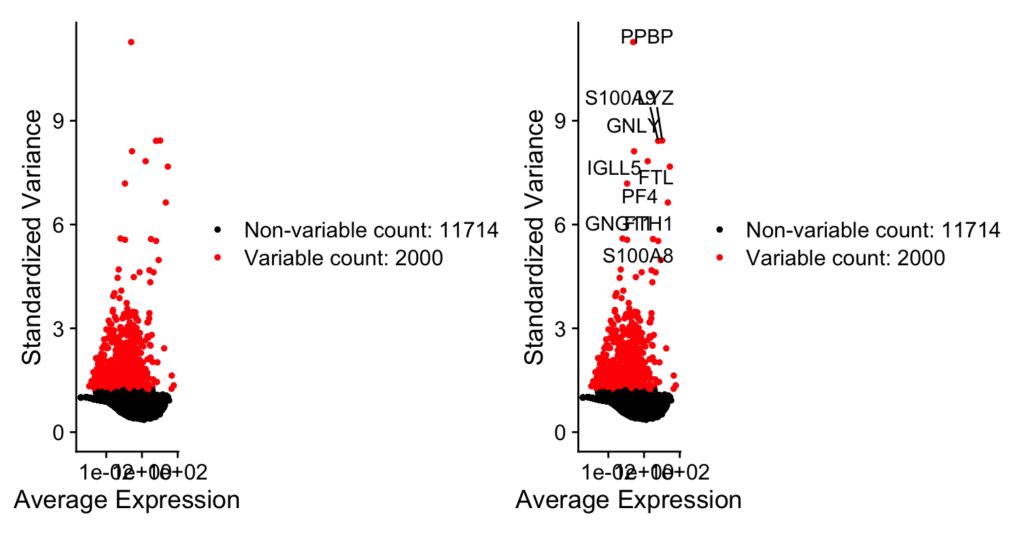
もしコマンド通りに叩いてもエラーが出る場合は…
コマンド通りに叩いてもエラーが出る場合は、pbmc に入る値が間違っています。エラーが出ても慌てずに、クオリティチェック→正規化…とコマンドを最初から叩き直してください。順番通りきちんと入力すればエラーなく通ります。
本記事では、一旦ここまでにしようと思います。
後半はこちらになりますのでぜひ挑戦してください!
最後に
いかがだったでしょうか。ハンズオンは折返しになります。次の記事も取り組んで一通りSeuratを使えるようにしてみましょう。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

