

この記事はGaussianを使った構造最適化計算で出てくるエラーへの対処方法を解説します。エラーに対処する流れの説明から、Gaussviewだけでなくテキストエディタを用いたエラー確認方法、具体的な対処法まで扱います。初めて構造最適化を行う方や構造最適化が収束しない方でも、計算を収束させることが出来るようになっています!
【この記事のまとめ】
Gaussianを用いた構造最適化計算で頻発するエラー(L9999, L103, L502など)の原因を特定し、確実に計算を収束させるための実践的な対処法を解説します。
- エラー特定とLinkの仕組み:出力ファイル(.out/.log)末尾の「Link(lXXX)」番号から、計算のどのフェーズ(Overlay)で問題が起きたかを診断する方法を提示。
- 主要エラー別の具体的解決策:初期構造やキーワードミス(L1, L101)、分子の直線状配置(L103)、SCF未収束(L502)、最適化未完了(L9999)など、状況に応じたオプション設定(
nosymm,scf=xqc,opt=MaxCycle等)を詳述。 - 再計算と効率化のテクニック:計算を途中から再開する
opt=restartや、収束状況を可視化する「Optimization plot」の確認方法など、試行錯誤の時間を短縮するノウハウを公開。
この記事を読むことで、複雑なエラーメッセージに惑わされることなく、論理的なステップで問題を解決し、構造最適化を成功に導くスキルが身につきます。
Windows 11(22H2), Gaussian16、Avogadro 1.2.0


Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!


初めに
構造最適化の計算では、収束しやすいGaussianを使っても、エラーに出会うことがよくあります。初学者の場合、頻繁にエラーが出てしまい、心が折れそうになる時があるでしょう。しかし、一つずつエラーに対処していくことで、徐々にエラーが減っていき、出会ってもすぐに対処できるようになります。めげずにエラー向き合うことが収束への近道です!
ではエラーの対処法について解説していきます!
エラーに対処し構造最適化を収束させる流れ
計算を実行した後に起こるエラーには以下の流れで対応していきます。
- 出力ファイルからエラーの種類を確認する
- エラーの種類から対処法を探し、入力ファイルの書き換えを行う
- 修正した入力ファイルを使って再度Gaussianを用いて計算する
Gaussianのエラーを確認するには?
まず、Gaussianで行った計算のエラーを確認する方法から解説していきます。
計算エラーの確認方法
- 計算のエラーは計算を実行した後に出てくる出力 ファイル(”.out” or “.log”)をエディタアプリ(サクラエディタやメモ帳など)で開きます。
- 開いたファイルの最後の数行を確認。
- 下図のようなエラー文が出ていればエラー終了しています。
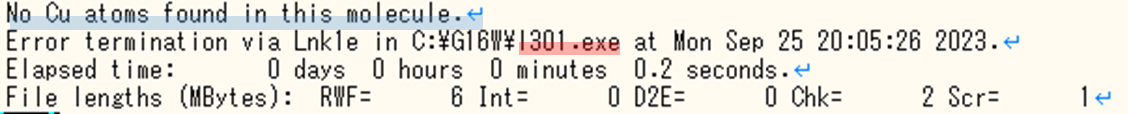
エラー終了している出力ファイル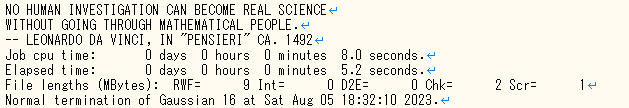 計算が最後まで終了したときの出力ファイル
計算が最後まで終了したときの出力ファイル - エラーの種類を確認。 エラー終了している場合、エラーの種類は赤色でハイライトした部分(今回は”l301”)を確認することで判別できます。青色でハイライトした部分はなぜエラーを起こしたのかを説明してくれるエラー文であり、今回であれば ”No Cu atoms found in this molecule.”、つまりCu原子が分子内に含まれていないことによるエラーだと分かります。
GaussViewで出てくるエラーメッセージ
因みに、エラー終了しているファイルをGaussViewで開くと以下のようなメッセージが出てきます。この場合も同じように出力ファイルをエディタアプリで開きエラーの種類を確認してください
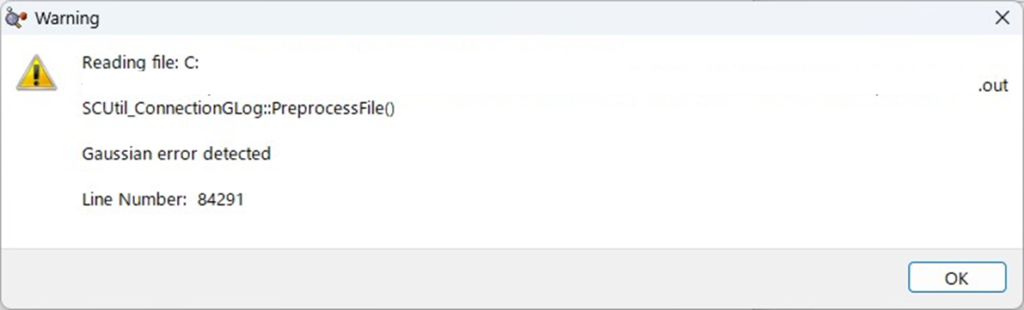
エラーの種類とLink
エラーの種類は今回の”l301”以外にもl0やl101, l9999など様々な種類が存在します。
これらは計算中の各ステップを表しており、”l301”と表示されている場合、”l301”を実行中に不具合が起こって計算が終了したことを示しています。因みに”l”は”Link”と呼ばれるGaussianの小プロセスを表しており、”l301”はLink301を実行している意味です。
では、各小ステップが何を実行しているのかを解説し、エラーの原因を特定していきます。
Gaussianの構造最適化計算の流れ
先ほど述べたようにLinkとは小プロセスのことであり、百の位が大プロセスを表しています。
大プロセスはOverlayと呼ばれ、Gaussianの計算はOverlay 0から始まりOverlay 99で終了します。
Overlayは基底状態の構造最適化、振動数計算、電荷計算等を行う場合、以下の図のような番号に役割が割り当てられています。
そのため、エラー解決の際、どのOverlayやLinkでエラーが発生したかを理解することが重要であり、それによって問題解決が容易になることがあります。
エラーに出会った際は、まずどのOverlay(エラー番号の百の位)を確かめましょう!

Gaussianの構造最適化でよくあるエラー
最後に、Gaussianを用いて構造最適化計算したときによく出会う各エラーについて、エラーの種類ごとに対処法を解説していきます。
読者が直面しているエラーの番号が、解説しているエラーの番号と異なっていても、同じOverlayのエラーであれば同様に解決できる場合があるため、参考にしてみてください!
l1
内容:
キーワードの解釈、初期化、開始処理
主な原因 :
・ファイルの⼊⼒形式が間違っている Link 0コマンドの記述が間違っている
・Gaussianキーワードの記述方法が間違っている など
対処⽅法:
百の位がないL1はOverlay 0で起こるエラーです。原因は主に入力ファイル内の作成・入力ミスによるものが多い為、再度入力ファイル内を確認しましょう!L1エラーが起こらないようにするための入力ファイルチェックリストを載せておきます。参考にして見てください!
エラーを少なくするための入力ファイル最終チェックリスト
- [ ] 入力ファイルの最後に空行はあるか?
- [ ] 各セクションの区切り空行は入っているか?
- [ ] 全角文字が入っていないか
- [ ] 使用するメモリ量・プロセッサ数は適切か
- [ ] Gaussianキーワードは正しいスペルか?(汎関数やopt, freqなど)
- [ ] チェックポイントファイル(.chk)の名前に特殊文字([,][.][[\][:][¥]など)が入っていないか
- [ ] 電荷とスピン多重度は正確か?
- [ ] 分子構造指定は適切か?
l101
内容:
タイトルと分子指定の読み込み
主な原因:
・原⼦の座標指定部分に不要な⽂字が含まれている
・電荷・スピン多重度の設定ミス 空行が不足している ⼊⼒が不⾜している
など
対処⽅法:
こちらはOverlay1でよく起こるエラーです。入力ファイルの④分子指定セクションの再確認を行いましょう。よくあるエラーの要因として、電荷とスピン多重度の値の間にスペースを入れ忘れていたり、スピン多重度が0になっている場合があります。また、分子指定セクション前後の空行や、Z-matrix座標で記述した際の座標後に入れる空行を忘れてもこのエラーが出ます。こちらもL1のエラーと同じように上記のチェックリストを確認してください。
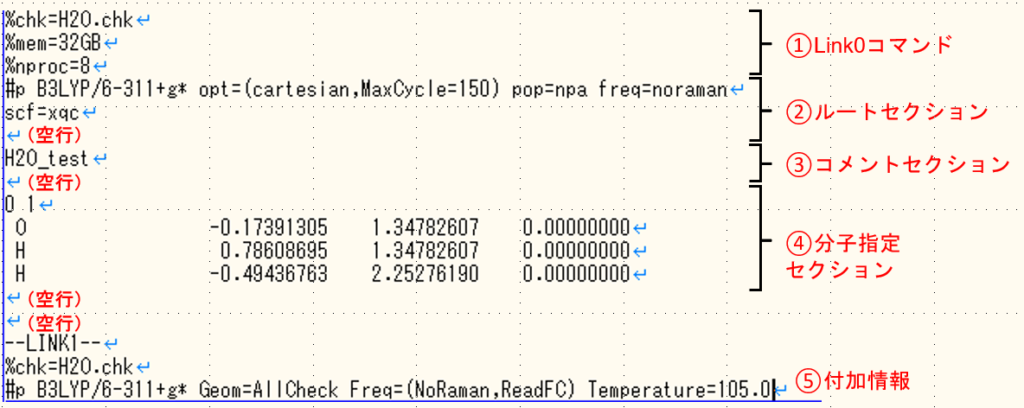
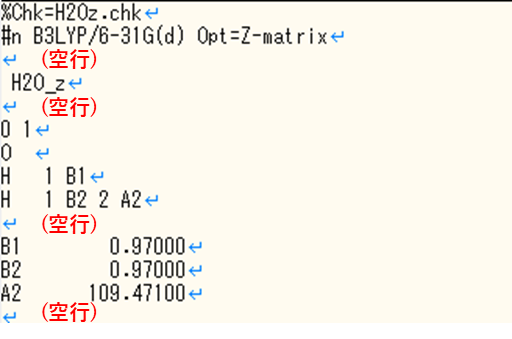
l103
内容:
構造最適化中のエラー
主な原因:
構造最適化中に原⼦が3つ直線状に並んだ、など
対処⽅法 :
こちらもOverlay1でよく起こるエラーです。表⽰されている原⼦をわずかにずらし、直線に並ばないようにして構造最適化を再開する方法が解決方法として考えられます。また、よくこのエラーに出会う方はnosymmのオプションを付けることで解決できる場合があります。 Gaussianは通常、分子の対称性を固定して計算が実行されますが、対称性が固定されることによって計算が収束しない可能性があります。nosymmは、指定した構造の対称性を無視して計算を回すオプションであり、対称性を固定して計算するより計算時間が掛かってしまいますが、エラーを解決できる可能性があります。nosymmの指定方法は以下のようにルートセクションに記載してください。(4行目)
nosymmを設定した入力ファイル例
%chk=H2O.chk
%mem=32GB
%nproc=8
#p HF/STO-3G opt nosymm
scf=xqc
H2O_test
0 1 O -0.17391305 1.34782607 0.00000000 H 0.78608695 1.34782607 0.00000000 H -0.49436763 2.25276190 0.00000000l202
内容:
原⼦座標の再配向、対称性の制御、変数のチェック
主な原因:
座標が間違っている
など
対処⽅法:
原⼦が重なっているなど、明らかにおかしい座標の指定をしていないか確認しましょう!入力ファイルをGaussviewやAvogadroなどの分子描写ソフトで表示し、表示された分子の座標を確認してください。座標を修正後、分子座標を出力してください。
Avogadroを使った分子座標の出力方法
1.エラーが出た入力ファイルをAvogadroにドラッグ&ドロップしてください。
2.以下の手順で重なった分子座標を修正してください。以下Avogadroでで修正している画像を載せています。
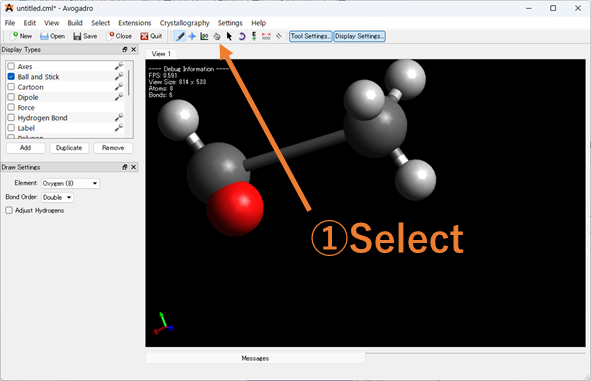
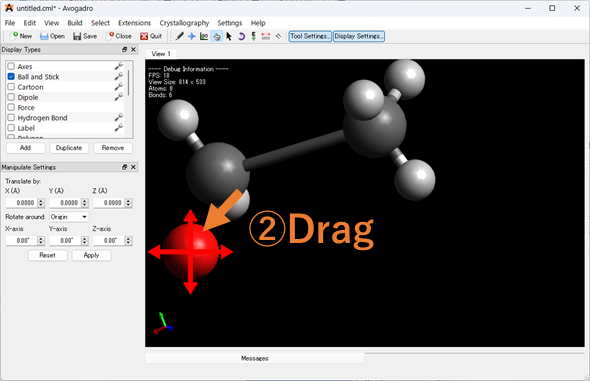
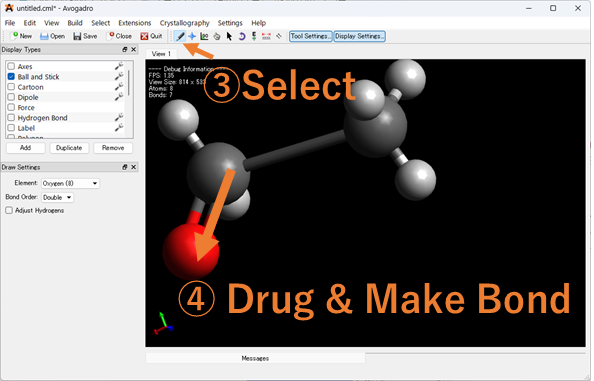
3.座標を出力していきます。Extension > Gaussian > Generate の順に入力ファイルを出力し、そこから分子座標を抜きだして再度計算を実行してください。
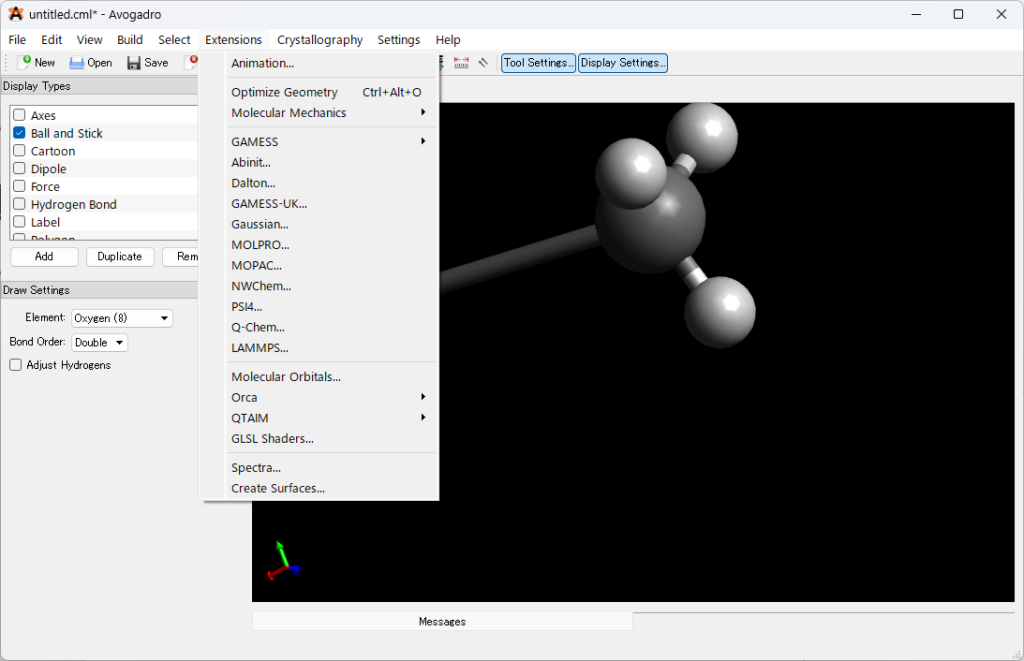
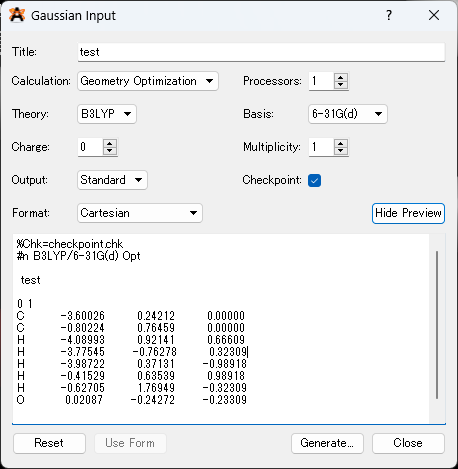
Gaussviewを使った出力方法
分子座標を修正後、分子を表示している画面で右クリックを押し、Calculate>Gaussian Calculation setup..>submitの順に出力してください。出力された入力ファイルから分子座標を引き出し、これまでの入力ファイルの分子座標を置換して、計算を再開しましょう!
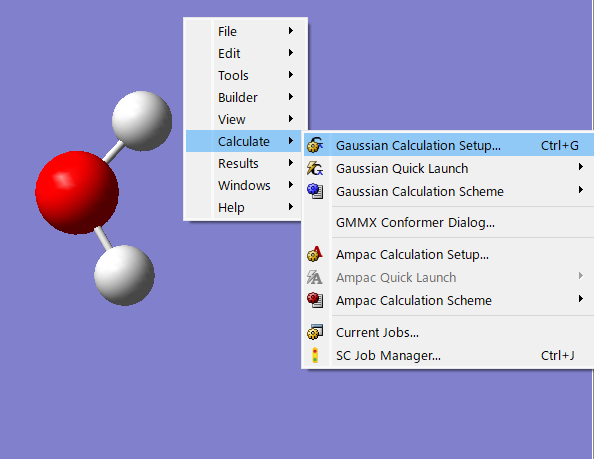
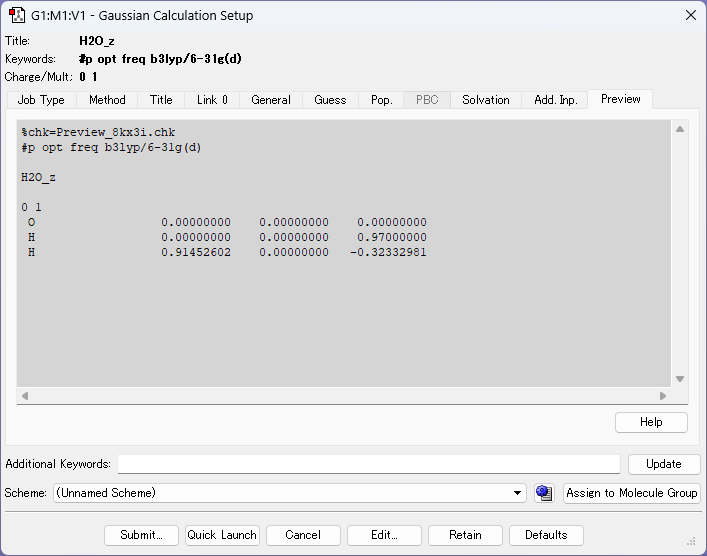
l301
内容:
基底関数情報の⽣成
主な原因:
・電荷とスピン多重度の値が不適切 基底関数の不正指定
・電荷とスピン多重度の不整合 Guess=Readで、存在しないchkファイルの読み込み
など
対処⽅法:
電荷もしくはスピン多重度が原因の場合
値を適切なものに変更して再度計算を実行しましょう。スピン多重度は先行研究などを参考に決定すると良いでしょう。このエラーの場合は以下のようにError terminationメッセージのすぐ上のエラー文がヒントになることが多いです。今回であれば電荷とスピン多重度が不適合であると記載されています。

各元素に基底関数を適用する場合
基底関数の名前を正確に書くことや、入力ファイルの書き方を見直しましょう。各元素に基底関数を設定する場合に特に起こるので、以下を参考にして入力ファイルを編集してください。
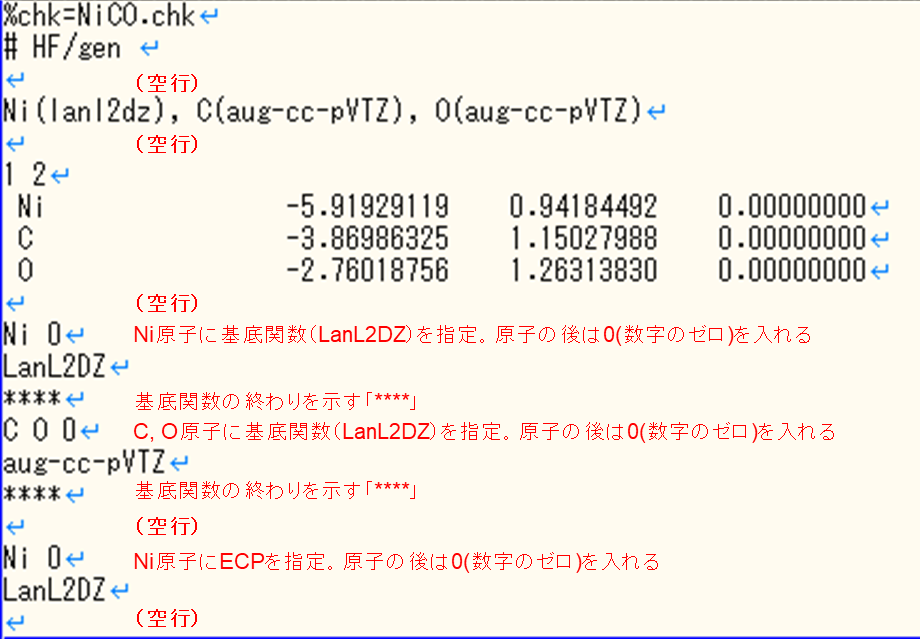
l502
内容:
SCF⽅程式の反復的解法
主な原因:
SCF計算が規定の回数内に収束しなかったなど
対処⽅法:
SCF手続きは、物質の性質がちゃんと計算できるようになるまで、何回も繰り返します。その状況を見るのにRMSDPという値を使います。これが収束していない場合エラーが出ます。
Overlay5のエラーは、現象としては一種類ですが、根本的な原因は様々あり対処が難しいエラーです。以下に代表的な対処法を載せておきます。この中から順に試してみましょう!
エネルギーが収束しない場合の対処法
- SCF手続きのアルゴリズムを変えて、SCF=Fermi、QC、XQCの中から1つを試す。
- 低精度な基底関数の計算結果を最初に使う。
- SCF=(MaxCyc=N) の N の値を増やす(デフォルトは N=128)。
- SCF=(Conver=N) の N の値を減らす(RMSDPの値が10−8や10−7の辺りを行ったり来たりしている場合は、Conver=7やConver=6を指定して、収束閾値を10−7や10−6に設定してやれば、それらの値でも直ちに収束と見做されます。)
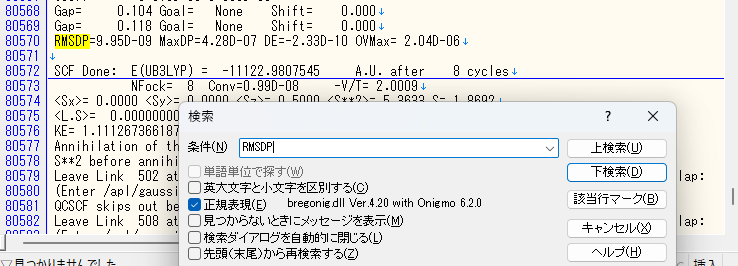
SCFオプションにCoverとXQCを設定した入力ファイル例
%chk=H2O.chk
%mem=32GB
%nproc=8
#p B3LYP/6-311+g* opt scf=(Conver=6, xqc)
H2O_test
0 1 O -0.17391305 1.34782607 1.00000000 H 0.78608695 1.34782607 0.00000000 H -0.49436763 2.25276190 0.00000000それでも収束しない場合は以下の選択肢も検討してみてください。
- Unrestricted (U)-SCF法を試してみる。
- (N+1)電子閉殻分子の計算結果を最初の情報に使う。
- 占有軌道と非占有軌道を適切に入れ替えて、最初の情報に使う。
- そもそもHF/DFT計算では計算できない
l9999
内容:
計算結果のまとめの出⼒
主な原因:
構造最適化が終了しなかった、など
対処⽅法:
対策の種類
Opt=(MaxCycle=XXX, MaxStep=X)を指定しましょう(Xは数字)。
| エネルギーは減少しているが時間がかかっている場合 | 同じ構造が繰り返し振動している場合 | |
| 指定オプション | MaxCycle | MaxStep |
| デフォルト | MaxCycleのデフォルトは系によって変わってきます。出力ファイルないのCycle数を確認し、その数に任意の数を加えてください。 | MaxStepのデフォルトは30なので設定する際は10ずつ減らしていきましょう。 |
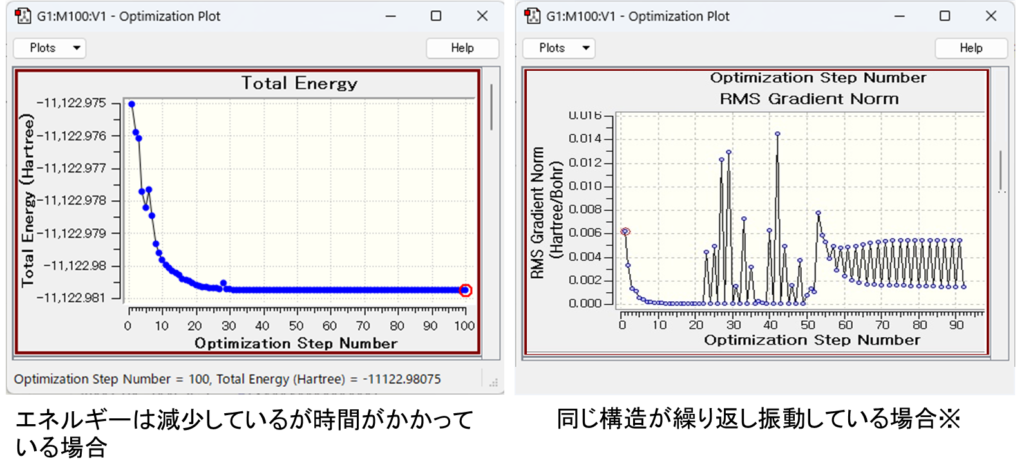
Gaussviewで構造最適化の結果(Optimaizetion plot)を調べて、同じ構造が繰り返し振動しているようならMaxStepを、エネルギーは減少しているが時間がかかっている場合はMaxCycleを指定するとうまくいくことが多いです。
Optimaizetion plotの確認方法はGaussviewとエディタソフトを用いて下で解説します。 MaxStepのデフォルトは30なので設定する際は10ずつ減らしていきましょう。
MaxCycleのデフォルトは系によって変わってきます。出力ファイルないのCycle数を確認し、その数に任意の数を加えてください。時間が掛かっても収束させたい場合は100を設定しても大丈夫です。
また、チェックポイントファイルファイル(%chk=×××.chk)はそのままに、再度計算を回すときはgeom=allcheckを設定しましょう。こうすることで、最後のステップの分子構造から計算を再開することが出来ます。
%chk=H2O.chk
%mem=32GB
%nproc=8
#p B3LYP/6-311+g* opt=(MaxCycle=100, MaxStep=20) geom=allcheck
H2O_test
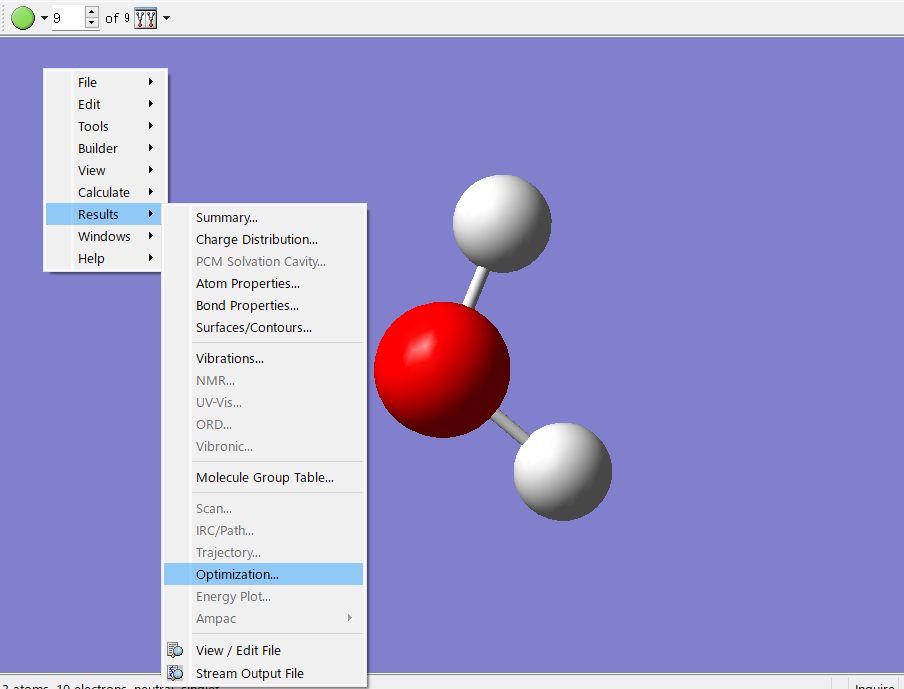
0 1 O -0.17391305 1.34782607 1.00000000 H 0.78608695 1.34782607 0.00000000 H -0.49436763 2.25276190 0.00000000Gaussviewを用いたOptimaizetion plotの確認方法
エラーが出た出力ファイルをGaussviewで開きます。その後、分子が表示された画面で右クリック>Result>Optimalization..を選択することでOptimalization plot が得られます。
エディタソフトを用いたOptimaizetion確認方法
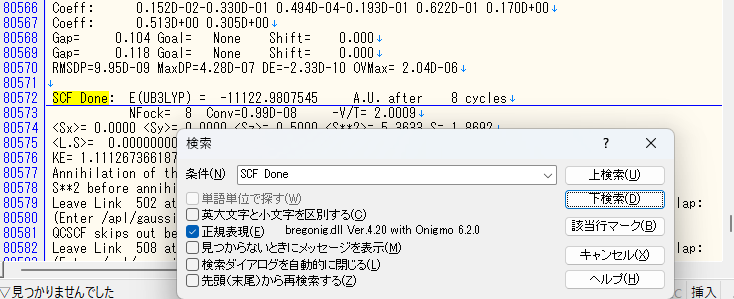
Gaussviewを使わず、エディタでOptimalizationを確認することもできます。出力ファイルをエディタソフトで開いた後、”control+F”キーを押してSCF Doneの文字列を検索してください。右に出ているE(UBLYP)= の後がエネルギーです。このエネルギーが下検索していくにつれて収束して居ればMaxcycleを、繰り返しがみられるようであればMaxstepを設定しましょう!
突然計算が終了している
主な原因:
ジョブの上限時間に計算が終了しなかった、gjfファイルが読み込めなかったなど
対処⽅法:
この場合は主に計算機の問題であることが多いです。計算機に時間を指定して計算している場合は計算を流す際に、現在よりも⻑い時間を設定しましょう。また、計算機側がスペースなどが名前に入った入力ファイルを読み取れず、このようなエラーが出るときがあります。これもL1エラーと同じように入力ファイルの最終チェックをしましょう!
また、計算を再開するときはOptのrestartのオプションを活用しましょう!指定したチェックポイントファイルから自動的に読み取り、計算を再開してくれます。設定は以下のようにしてください。ルートセクションにを設定する以外は最初の計算で使用した入力ファイルのままで大丈夫です!
restartを指定するときの入力ファイルの例
%chk=H2O.chk
%mem=32GB
%nproc=8
#p B3LYP/6-311+g* opt=(MaxCycle=100, restart)
H2O_test
0 1 O -0.17391305 1.34782607 1.00000000 H 0.78608695 1.34782607 0.00000000 H -0.49436763 2.25276190 0.00000000最後に
いかがでしょうか。Gaussianのエラーは熟練者でも躓くことがよくあります。なぜエラーが起こったのかを考え、周りの研究者と議論しながら解決してみてください。その際にこの記事が役に立てば幸いです。
参考
[1] 西長亨、本田康 「有機化学者のための量子化学計算入門 Gaussianの基本と有効利用のヒント」 第3・4・5章 P. 32-91[2] 平尾公彦、武次徹也「新版 すぐできる 量子化学計算ビギナーズマニュアル」p. 92[3]https://www.researchgate.net/post/What_are_possible_solutions_when_the_during_geometry_optimization_the_energy_oscillates_as_shown_in_attachment
Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

