
本記事では、Avogadroを使ってGaussianの入力ファイルを簡単に作成する手順を解説します。分子座標の作り方に困っている方や、Avogadroについて知らない方でも、すぐに使えるように解説していきます!
今回使用するソフトウェアであるGaussianおよびAvogadroは以下の目的で利用しています。
Avogadro : 計算を実行する入力ファイルの分子座標を作成、振動数計算の虚振動を確認
Gaussian : 構造最適化及び振動数計算の実行、計算結果の出力
Windows 11(22H2), Gaussian16、Avogadro 1.2.0

Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!
Avogadroとは?

Avogadroは、分子の3D構造をモデリングし、分子座標を作成および視覚化するための無料で利用できるソフトウェアです。主な特徴は以下の通りです:
- 分子モデリング: Avogadroは、分子の原子や結合を直感的に配置し、3Dモデルの分子座標を構築するのに役立ちます。また、小さな分子だけでなく、タンパク質や結晶構造などの大きな系もモデリングすることができます。
- 多くのフォーマットのサポート: Avogadroは、多くの分子ファイルフォーマットをサポートしており、他の化学ソフトウェア(Gaussian やGAMESS等)とデータを簡単に共有できます。これにより、異なるソフトウェア間でデータを円滑にやり取りできます。
- オープンソース: Avogadroはオープンソースソフトウェアで、無料で利用できます。また、コミュニティによって開発され、継続的に改良が行われています。
- プラグインサポート: Avogadroはプラグインアーキテクチャを採用しており、さまざまな追加機能を統合できます。これにより、ユーザーは必要に応じてソフトウェアの機能を拡張できます。
総括すると、Avogadroは化学者や研究者にとって非常に役立つツールであり、分子のモデリングと計算に関する多くの機能を提供しています。特に高価なGaussviewと異なり、無料で利用できることが大きな特徴です。
今回はこのAvogadroを用いて簡単にGaussianの入力ファイルを作成していきます!
Avogadroのインストール
早速、Avogadroをインストールしていきましょう!
Avogadroは以下の公式サイトでダウンロードすることが出来ます。
Avogadroのダウンロードページを開くと以下のようなページに遷移するので、タブ内中央の”Download”をクリックしてください。自動的にダウンロードが開始されます。
次に、ダウンロードした”Avogadro-1.2.0n-win32.exe”ファイルを開いてください。ここの1.2.0の部分はAvogadroのバージョンを示している為、変わる可能性があります。開くと管理者権限を問われる画面になりますので、許可してください。
許可するとセットアップ画面が現れます。以下の手順に従ってセットアップを進めてください。
①下の次へ(N)を押してください。
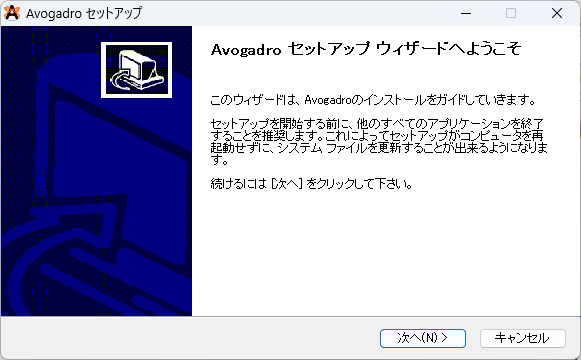
②ライセンス契約書に同意します。
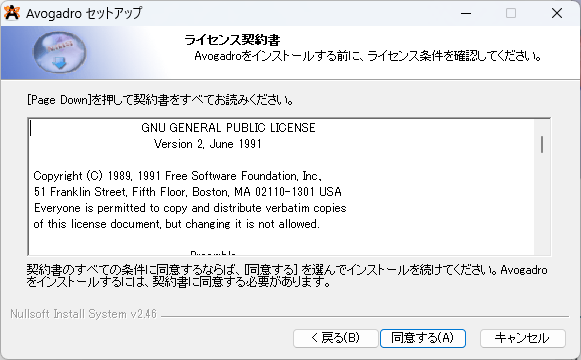
③インストールオプションを選択します。ここではAvogadroのPathを自動でPCに設定してくれます。画像のように選択してください。ログインしているユーザーアカウントのみでPathを設定する場合は、3つ目を選択しましょう。また、すぐ使えるようにCreate Avogadro Desktop icon を選択しておきましょう。
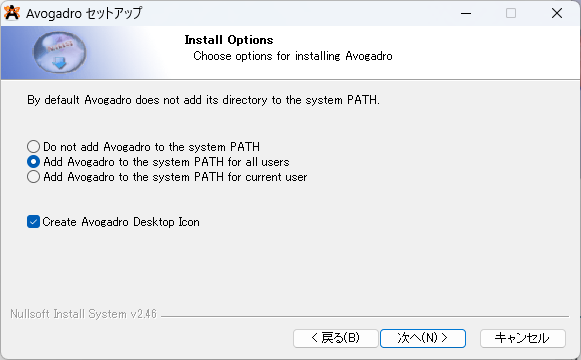
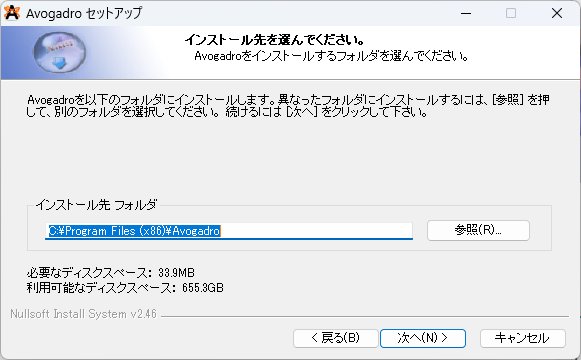
④最後にインストールするフォルダを選択してください。選択後、次へ(N)を押すとインストールが始まります。
Avogadroを使って入力ファイルを作成する方法
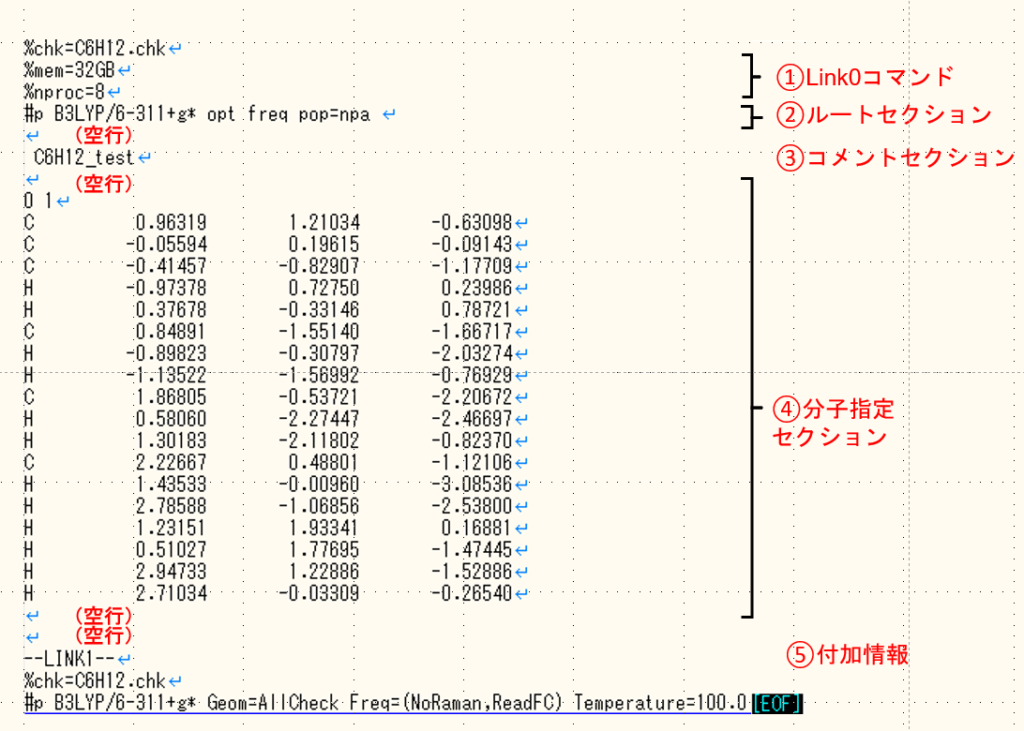
Avogadroを使って、GaussianでC6H12を構造最適化と振動数計算を行うように指示する入力ファイルを上の画像のように作成していきます。
Gaussianによる構造最適化の計算では、初期構造が計算コストに大きく影響します。そのため出来るだけ安定構造に近い構造を作ることが効率良く構造最適化を進めるためのテクニックになります。
ここでは安定構造近い分子座標をAvogadroを用いて作成し、Gaussianの入力ファイルを作成していきます。
1.Avogadroを用いた分子座標の作り方
上の画像を参考に、以下の手順で作っていきましょう!
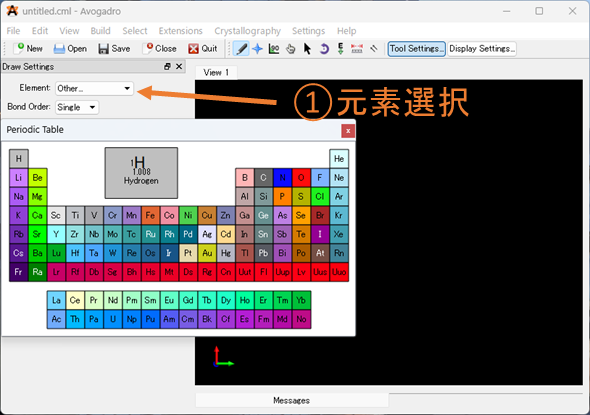
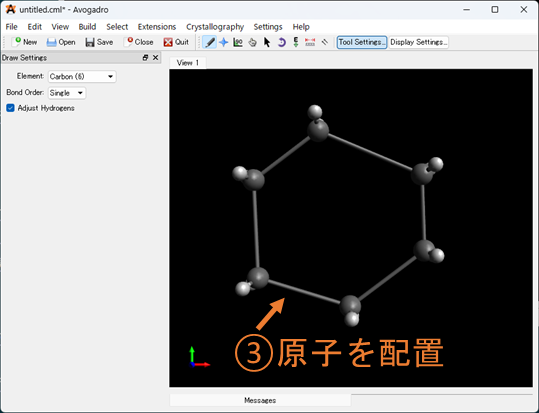
①元素選択: 追加したい元素を選択してください。デフォルトは炭素になっています。金属などを選択したいときはOtherを選択すると、周期表が出てくるので、そこから選択して下さい。
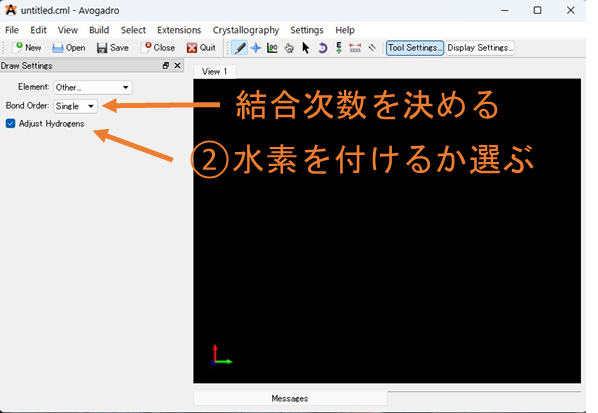
②水素を付けるか選ぶ: 分子を作成するときに自動で水素を付けるか選択することが出来ます。また、水素が必要ないときは、Adjust hydrogen をONにしたまま任意の原子を選択することで削除することもできます。
③原子を配置: 黒い領域をクリックすると原子が配置されます。配置された原子をクリックし、任意の位置にドラッグすることで選択した原子から結合を伸ばした場所に原子を配置することが出来ます。結合形式はデフォルトでは単結合ですが、②の上にBond order があり、ここを任意の結合様式を選択し、分子の結合をクリックすることで単結合から結合次数を変更することが出来ます。
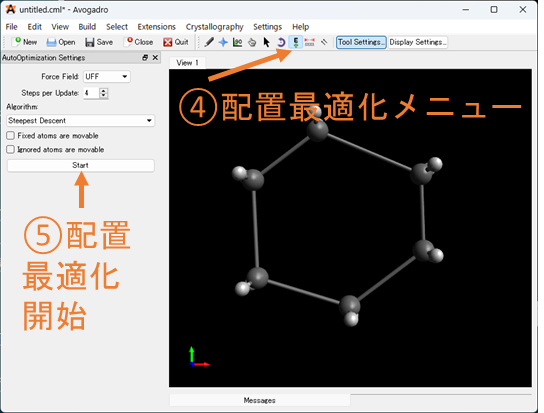
④⑤原子の配置を最適化する: 手動で原子が配置出来たら④の配置最適化メニューを選択し、⑤の配置最適化を開始してみてください。ここでは、簡単な力場を用いて分子動力学シュミレーションを行い、座標を修正してくれます。これをしておくことで計算時間が減少することがあるため、やっておきましょう!また、配置最適化メニューの2つ左の![]() を選択し、原子を動かす子ことで構造異性体を作る事もできます(C6H12の場合、構造異性体である舟形配座を作成できる)。遷移状態など任意の構造を作るときにも役に立ちます。
を選択し、原子を動かす子ことで構造異性体を作る事もできます(C6H12の場合、構造異性体である舟形配座を作成できる)。遷移状態など任意の構造を作るときにも役に立ちます。
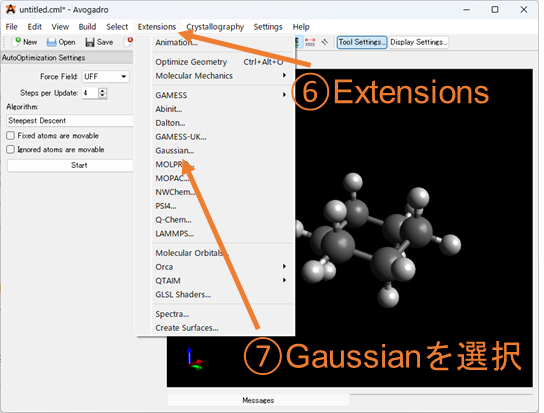
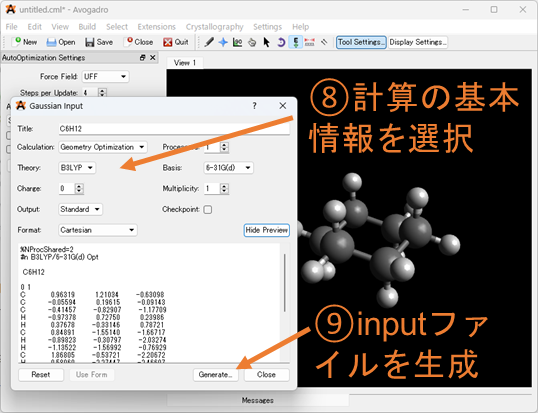
⑥⑦⑧座標の出力形式を選択する: ⑥のExtensionsを選択し、Gaussianの入力(.gjf or .com)ファイルの形式に沿った座標のファイルを生成していきます。⑦でGaussianを選択し、⑧で汎関数や基底関数、電荷・スピン多重度の設定を行ってください。ここで選択できない汎関数や基底関数はテキストエディタで編集してください。特にOptとFreqの同時に選択することが出来ない為、構造最適化+振動数計算を行う場合は編集する必要があります。
⑨入力(Input)ファイルの作成: 最後にファイルを出力していきます。出力は”.com”の拡張子で作成されます。研究室などのルールで”.gjf”の拡張子を使用している場合は後から編集してください。
結果こちらのようなファイルを得ることが出来ました。

今回はAvogadroを用いて手動で原子を配置しましたが、2Dの分子描画ソフト(Molvewなど)で書いたMOLファイル(拡張子.mol)やX線構造解析の結果から得られた座標、SMILES記法などを読み込ませることも可能です。
入出力ファイルお勧めエディタ
因みに入力ファイルの編集にはサクラエディタがお勧めです。
メモ帳等でも作成できますが、サクラエディタは分子座標の置換などが簡単にできます→こちらからダウンロード
2. 計算条件の指定
次に計算条件を編集していきます。特にAvogadro上では構造最適化(Opt)と振動数計算(Freq)を両方選択するなど、細かい設定を記入することが出来ない為、以下のように手動で記入しましょう。必要に応じて、基底関数や汎関数、計算条件などを編集してください。おすすめの条件等の説明はこちらの記事で紹介しています。
%chk=C6H12.chk
%mem=32GB
%nproc=8
#p B3LYP/6-311+g* opt freq pop=npa
C6H12_test
0 1
C 0.96319 1.21034 -0.63098
C -0.05594 0.19615 -0.09143
C -0.41457 -0.82907 -1.17709
H -0.97378 0.72750 0.23986
H 0.37678 -0.33146 0.78721
C 0.84891 -1.55140 -1.66717
H -0.89823 -0.30797 -2.03274
H -1.13522 -1.56992 -0.76929
C 1.86805 -0.53721 -2.20672
H 0.58060 -2.27447 -2.46697
H 1.30183 -2.11802 -0.82370
C 2.22667 0.48801 -1.12106
H 1.43533 -0.00960 -3.08536
H 2.78588 -1.06856 -2.53800
H 1.23151 1.93341 0.16881
H 0.51027 1.77695 -1.47445
H 2.94733 1.22886 -1.52886
H 2.71034 -0.03309 -0.26540
--LINK1--
%chk=C6H12.chk
#p B3LYP/6-311+g* Geom=AllCheck Freq=(NoRaman,ReadFC) Temperature=100.0
大量の計算を回す予定の方は、pythonなどで自動で記入できるプログラムを書くと便利です。
以上で入力ファイルの作成は終了です。
作った入力ファイルを、Gaussianを用いて計算してみて下さい。
Avogadroを使って振動数解析の結果を確認する方法
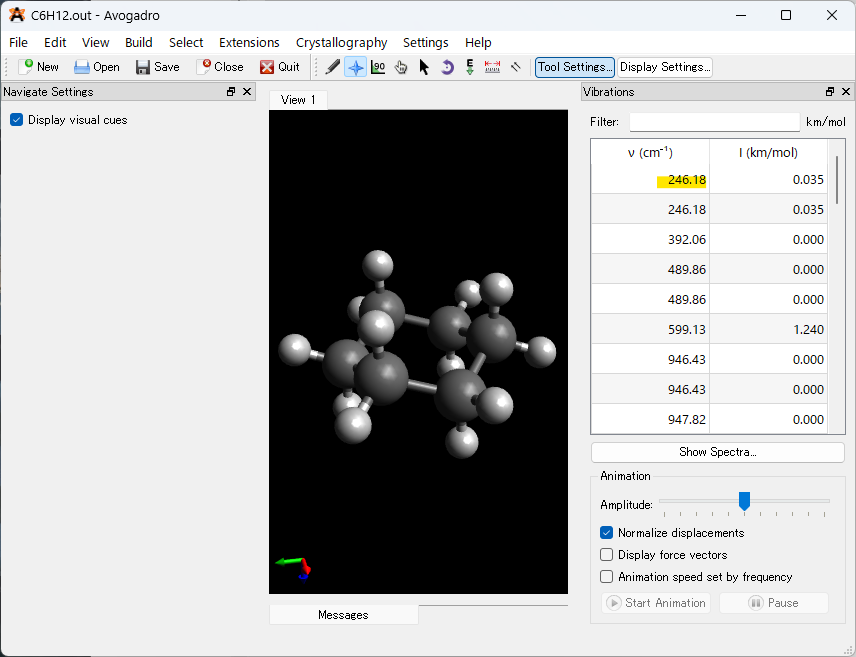
最後にGaussianを用いて計算終了後、作成された構造最適化・振動数計算の出力(.out or .log) ファイルからAvogadroを用いて安定構造と振動数計算の結果を確認していきます。
計算が終了した出力ファイルを左上のOpenボタンから開くと以下の図のような画面になります。
開くと画面左に3Dモデルの分子構造、画面の右に振動数が表示されます。Avogadroでは虚振動が現れているかをすぐに確認でき、黄色でハイライトしている部分が正の値を取っていれば安定構造で構造最適化計算が終了しています。
また、下のShow Spectraを押すことでスペクトルデータを表示させることが出来ます。
最後に
いかがでしょうか!今回紹介したように、Avogadroは力場計算を簡単に出来、簡単に入力ファイルを作成できる無料のソフトウェアなので、Gaussviewを使っている方もぜひ使ってみてください!
おまけ
エラーを少なくするための入力ファイル最終チェックリスト
- [ ] 入力ファイルの最後に空行はあるか?
- [ ] 各セクションの区切り空行は入っているか?
- [ ] 全角文字が入っていないか
- [ ] 使用するメモリ量・プロセッサ数は適切か
- [ ] Gaussianキーワードは正しいスペルか?(汎関数やopt, freqなど)
- [ ] チェックポイントファイル(.chk)の名前に特殊文字([,][.][[\][:][¥]など)が入っていないか
- [ ] 電荷とスピン多重度は正確か?
- [ ] 分子構造指定は適切か?
Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

