
本記事では、汎関数・基底関数の選び方から構造最適化(opt)・振動数計算(Freq)を行う際の入力ファイルの注意点を解説し、安定構造を得ることまで扱います。
初めてGaussian で構造最適化を行う方や振動数計算がよくわからない方、最近始めたばかりの方でも、簡単に分子の安定構造を計算できるようになっています!

Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!
構造最適化計算とは?
Gaussianを使用して行われる構造最適化は、分子の最安定な立体構造を見つけるために行われる計算手法です。
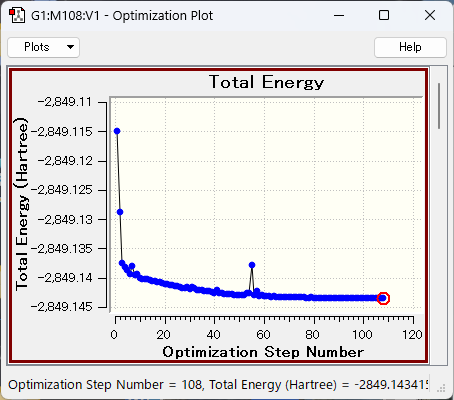
構造最適化では、図のように与えられた分子の原子の配置を変化させながら、エネルギー最小化の計算を行います。最適な分子構造を見つけるためには、分子のエネルギーを最小化するための最適な原子間距離や角度を少しずつ原子を動かしながら見つける必要があります。 分子のエネルギーを最小化する過程では、与えられた分子の初期構造を基にして分子のエネルギーや勾配(エネルギーの微分値)を計算し、最適化の収束基準が満たされるまで計算を繰り返します。
振動数計算と構造最適化計算を組み合わせて行う理由
実は、分子のエネルギー最小化を目指して(エネルギーの微分を計算して)原子の配置を調整する構造最適化計算だけでは分子が基底状態で安定かどうか確認できません。なぜなら、特定の原子配置がエネルギーの局所的な最小値(ポテンシャルの鞍点)に収束する可能性があるからです※。そこで、分子が真のエネルギー最小値(安定構造)に達しているかどうかを確認するために、振動数計算が必要になります。
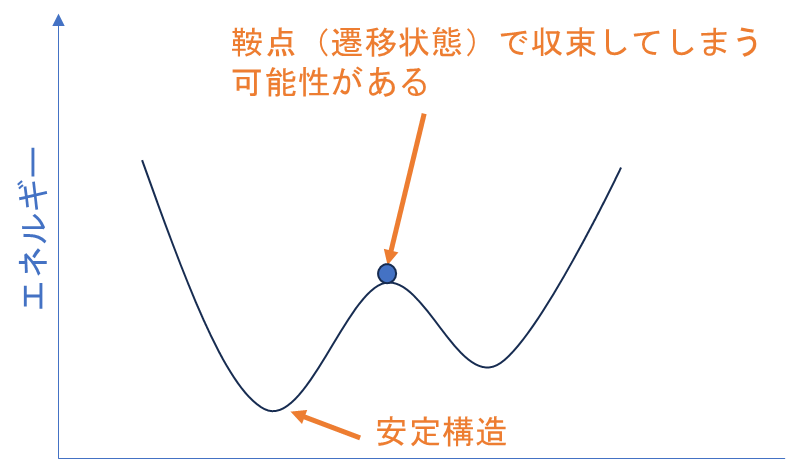
エネルギーの局所的な最小値かどうかは、振動数計算によって計算された振動数の符号を確認することで判断できます。真のエネルギー最小値では、分子の振動モードに対応する振動数はすべて正の値となります。一方、局所的な最小値では、1つ以上の虚振動が現れ、これにより1つ以上の振動数が負の値となります。これは最後のセクションで確認方法を解説します。
※Gaussianは、初期構造の分子対称性を固定することにより、計算時間を短縮します。ただし、この際、対称性が崩れる方向が安定であっても、原子配置を調整せずにそのまま計算を進め、収束してしまうことがあります。
構造最適化を行う手順
構造最適化の手順は大きく分けて以下の5つになります。
- 計算する分子の調査
- 計算手法の決定
- 入力ファイルの作成
- ジョブの投入
- 出力ファイルの確認・解析
では一つずつ解説していきます
1. 計算する分子の調査
まず、対象の分子について以下の項目を調べておきましょう。
①対象の分子、またはそれに類似した分子の先行研究(計算と実験を比べている研究が望ましい)
②対象分子の電荷、スピン多重度
③対象分子のおおよその分子座標(単結晶構造解析結果のcifファイルなどでも可能)
①は主に後の計算手法の決定に役立ちます。特に計算手法にDFTを用いる場合は、選ぶ汎関数によって結果が大きく変わる場合があるため、その系に合った汎関数を選ぶ時に役立ちます。またその際は、対象と近い系で実験結果と合う計算結果が出る汎関数を選ぶ事をお勧めします。
②は計算を回すのに必須になります。①で先行研究が見つかっている場合は、それを参考にしてみてください。
③については、小さな分子の座標は手計算で算出することもできますが、大きな分子の場合はGaussViewやAvogadroなどの分子描写ソフトで作成する事をお勧めします。
2. 計算レベルの決定
次に計算レベルをどう決めるかについて解説していきます。
計算レベルは計算手法と基底関数によって決まり、どの計算レベルを選ぶかで量子化学計算の計算精度と計算時間が決まります。基本的には計算精度と計算時間はトレードオフの関係(どちらかが大きくなるともう一方は小さくなる)の為、目的に応じて決定する必要があります。
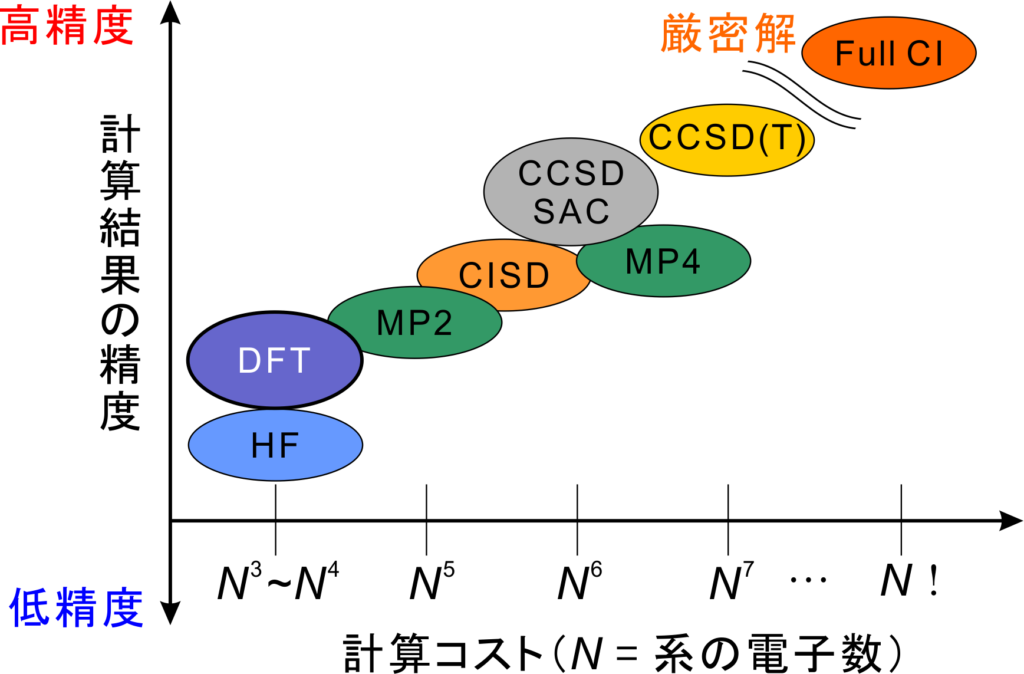
また、効率よく計算を進めるには計算手法と基底関数の質のバランスに注意することが必要です。例えば精度の高いCCSDなどの計算手法を使っても、基底関数がSTO-3Gなどの精度の低いものだとコスパが良い計算とは言えません。
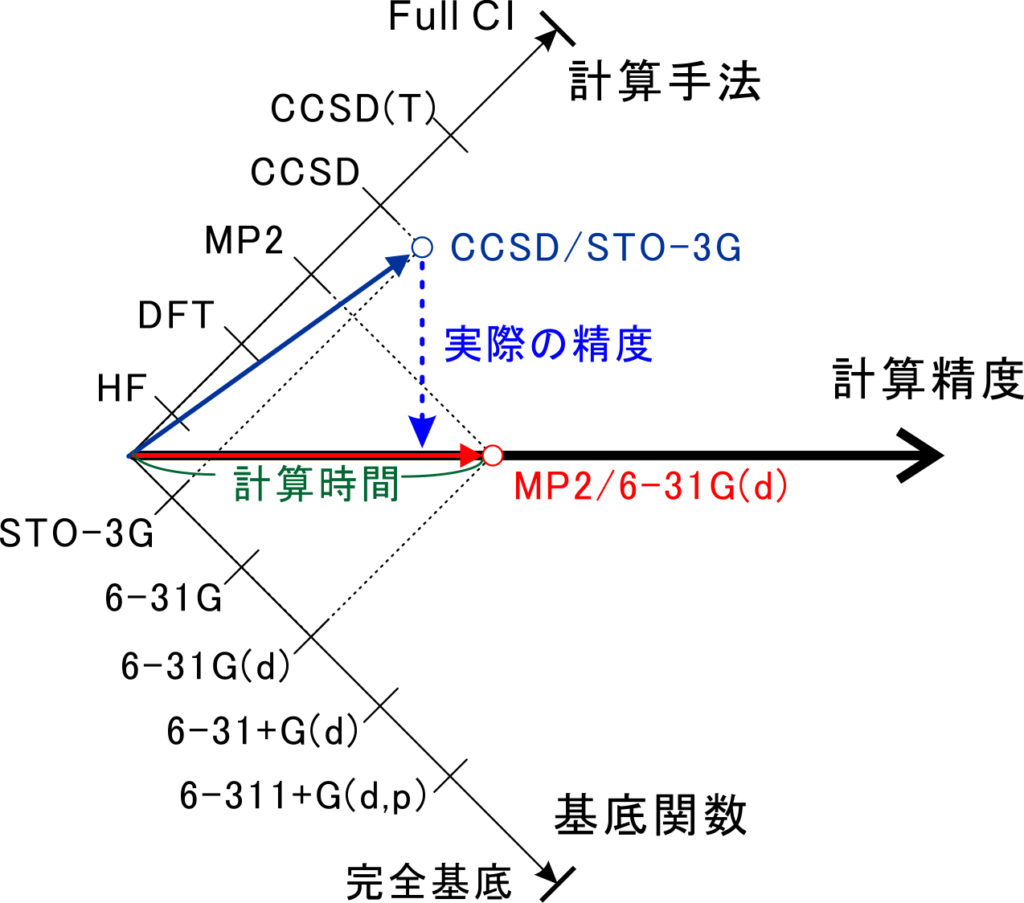
DFT計算をする時におすすめの計算レベル
しかし、まずは手っ取り早く始めたい方もいると思いますので、おすすめの計算レベルを載せておきます。計算する対象の分子が見つかっている方は先行研究で用いられている計算レベルを使うのもお勧めです!
とりあえずDFT計算を回したい人へのおすすめの汎関数&基底関数
| 対象の系 | おすすめの汎関数/基底関数 |
|---|---|
| 金属系触媒、クラスターなど | TPSSh/6-31G(d) |
| 水素結合がある場合 | M06-2X/6-31G(d,p) |
| その他 | B3LYP/6-31G(d) |
3. 入力(Input)ファイルの作成
Gaussianソフトで構造最適化を行うように指示する入力ファイルを各セクションごとに解説していきます。
入力ファイルの拡張子は”.gjf”と”.com”がありますが、どちらを使うかは同じ組織内で統一しておくことをお勧めします。
ファイルはテキストエディタを使用して閲覧・編集・作成することが出来ます。メモ帳等でも作成できますが、おすすめはサクラエディタです→こちらからダウンロード
サクラエディタは入力ファイル作成時に便利な機能を多く持っています。
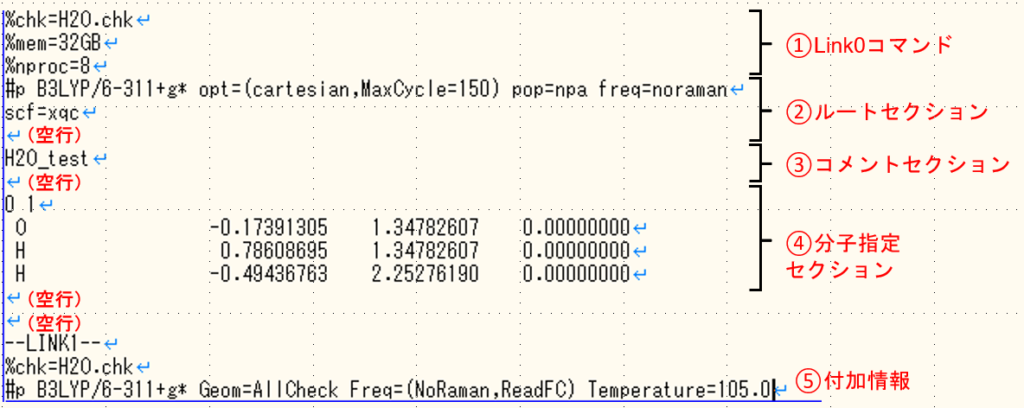
では、①〜⑤の各セクションの書き方については以下で紹介していきます!
①Link0セクション
%chk=H2O.chk
%mem=32GB
%nproc=8”%”を行頭に付け、1行に1項目ずつ記述します。一行目の.chkは中間ファイルのことで、分子軌道等を分子描写ソフトで可視化するときに使用するので作成することをお勧めします。ファイル名には分子の名前など分かりやすい名前がおすすめですが、”,”や”.”などの特殊文字を使用するとエラーが出る可能性があるので注意しましょう。
3行目のプロセッサ数は並列計算する場合に必要な記述です。また、同時に使用するメモリ量の記述が必要になります。プロセッサ数をして効率的に計算する場合は、目安としてプロセッサ数1に対してメモリ量を4GBとすると良いでしょう。
②ルートセクション
#p B3LYP/6-311+g* opt pop=npa freq
scf=xqc
“#”から始まるのがルートセクションです。ここでは、計算手法や基底関数、計算内容(構造最適化や振動数解析、エネルギー計算)を指定することが出来ます。何行かに渡って書き続けることも可能です。また、ルートセクションの最後に空行を入れることにより,ルートセクションの終わりを示します。空行は特に忘れやすく、エラーの原因になるので注意してください。以下に構造最適化でよく使う各Gaussianワードを解説しています。
| Gaussianワード | 内容 |
|---|---|
| p | 詳細な出力を行います。計算に掛かった実行時間などや,SCFの収束に関する情報が出力されます。 |
| B3LYP | 計算手法です。今回はDFTの一つであるB3LYP法を指定しています。 |
| 6-311+g* | 基底関数です。計算手法と基底関数は”/”で区切って指定します。(基底関数の「*」は「(d)」と同じ意味です) |
| Opt | 構造最適化を実行するキーワードです。このワードを入れるだけで構造最適化が実行されます。頭文字は小文字でも大丈夫です。 |
| Pop | 分子軌道の出力や、電子密度解析(population analysis)を実行するキーワードです。popは構造最適化計算では最初と最後の構造に対して行われます。=npaは電子密度解析の一つであるNatural Population Analysis (NPA)を指定するオプションです。 |
| Freq | 振動数計算を実行するキーワードです。対象分子の最安定構造を得たい場合はoptとセットで指定することが必要です。 |
③コメントセクション
H2O_test
複数行に渡るコメント入力が可能です。こちらもルートセクションと同様に、セクション終わりに空白が必要になります。
④分子情報セクション
0 1
O -0.17391305 1.34782607 0.00000000
H 0.78608695 1.34782607 0.00000000
H -0.49436763 2.25276190 0.00000000
ここでは分子の電荷、スピン多重度の後に、分子の初期構造の座標を記入します。
電荷・スピン多重度
電荷が左、スピン多重度が右になります。電荷は数字で表記され、0が中性、1が一価のカチオン、-1が一価のアニオンに対応しています。電荷・スピン多重度は先行研究などを参考に決定してください。この時、対称の分子系に対してありえない数値の電荷・スピン多重度を指定すると実行エラーが出るので注意してください。また、電荷とスピン多重度の間を半角スペースで区切ることも忘れないようにしましょう。
分子構造座標
今回はH2Oの分子構造座標を入力しています。構造最適化では、入力された構造を始点として、エネルギーが下がる方へ少しずつ座標を移動させ、最安定構造を探します。そのため、初期構造が安定構造に近ければ近いほど、計算が早く終わります。もし高い計算レベルを使用する際は、予め低い計算レベルで計算した分子座標を使用することをお勧めします。
今回は座標の記入方法としてデカルト座標(XYZ座標)を用いて記入しましたが、Z-Matrix座標を使って記入することもできます。(その場合、ルートセクションでopt=Z-Matrix を指定)
構造の選び方の例を以下に示します。
- X線構造解析や論文の補助情報(SI)で計算結果の情報が公開されている場合、その既存のデータを利用する。
- 有償のGaussviewや無料のviewerソフトウエア(Avogadro, winmostar)を使用して作成する。
Avogadroのダウンロードページを載せておきます
サクラエディタを使った分子座標編集テクニック
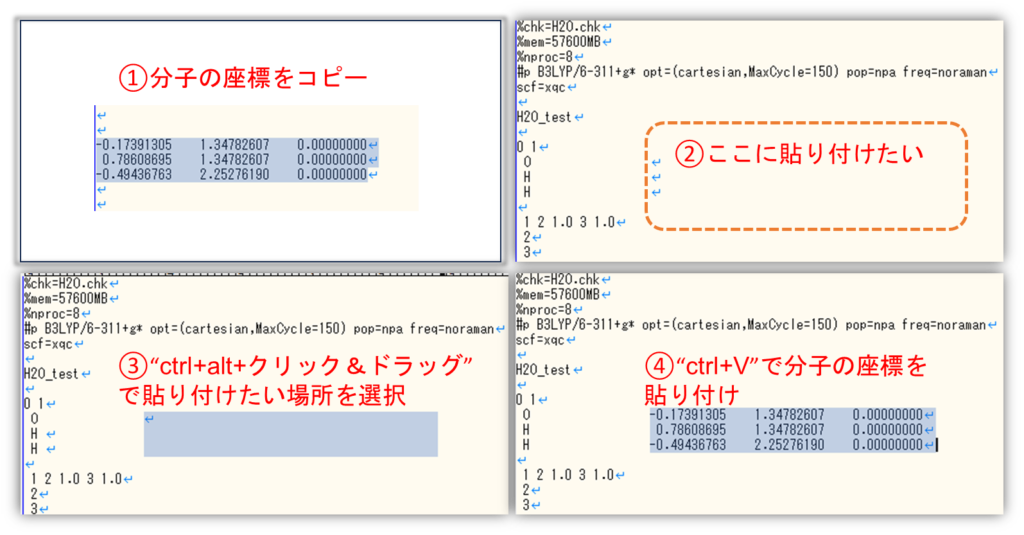
”ctrl+alt+選択したい箇所をドラッグ”することで分子座標だけを抜き出すことが出来ます。分子座標を置換したいときなど、入力ファイルの編集に非常に便利です。
⑤付加情報
--LINK1--
%chk=H2O.chk
#p B3LYP/6-311+g* Geom=AllCheck Freq=(NoRaman,ReadFC) Temperature=100.0
--LINK1--を付けると、上記の計算を行った後に追加で行う計算を指定することができます。例えば、通常設定では振動数計算によって得られるギブズエネルギーなどは298.150 Kにおけるエネルギーになりますが、場合によっては他の温度におけるエネルギーが必要になることがあります。そこで、LINK0で行った計算結果を基に再度温度を変えて振動数計算をすることで、指定した温度でギブズエネルギーなどを得ることができます。
Geom=AllCheckは上で行った計算結果が入っている.chkファイルから分子座標等を読み込んでいます。Freq は上記でも示した振動数計算のことで、ラマンデータが必要ないときは =(NoRaman,ReadFC)を付けておくと計算時間を短縮することが出来ます。Temperature=100.0は設定する温度(K)です。再度Freqを行う場合、同じ計算レベルを使用する必要があることに注意してください。
また、⑤を記入しない場合は、分子の座標を入力した後に2行以上のの空白が必要になることに注意してください。
4. ジョブの実行方法
入力ファイル(.gjfまたは.comファイル)が出来ればいよいよGaussianを使って計算していきましょう!
自身のPCに入っているWindows版のGaussianで実行する場合
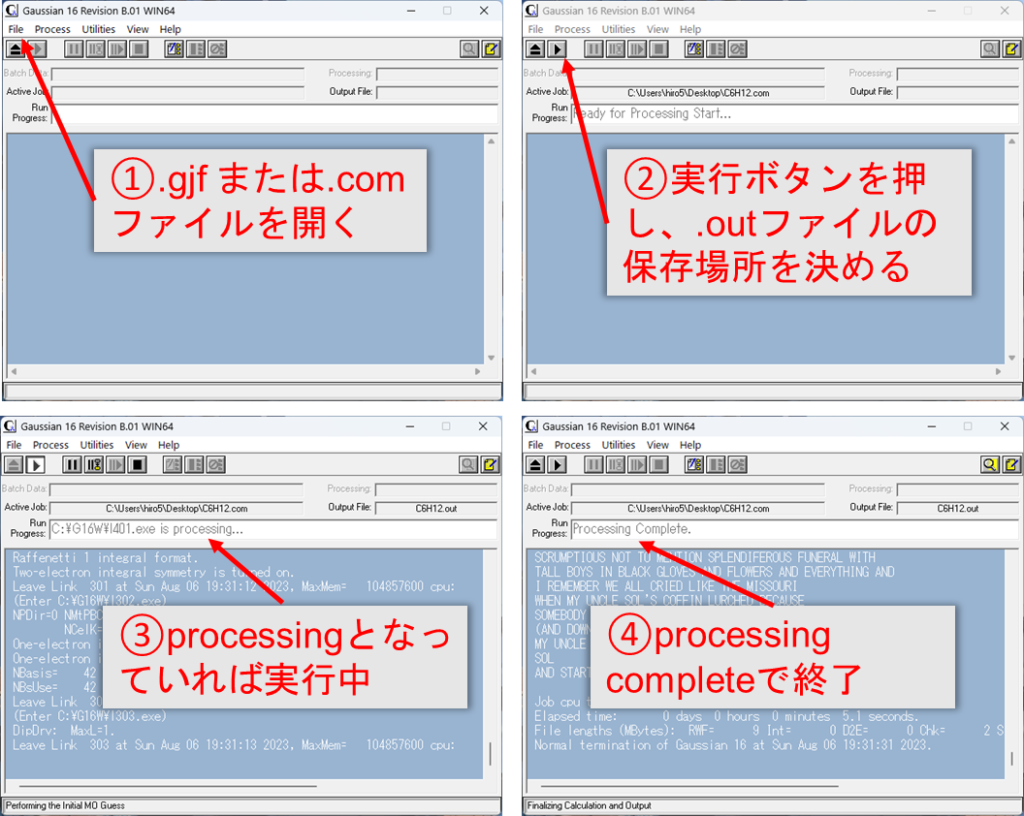
Windows版の場合、Gaussianのソフトウェアを開き、上記のようにファイルを開くorドラッグして開いて、実行ボタンを押します。出力される.outファイルを置く場所尋ねられるため、分かりやすいフォルダを指定しましょう。最後に正常に終了しているか確認すれば終了です。
ワークステーションなどのUnix/Linax版のGaussianで実行する場合
高速計算機など計算サーバーで実行する場合は以下の手順で行います。以下は、複数の人がそのサーバを利用し、作業用のデレクトリが割り当てられている事を想定しています。
- WinSCP(入出力ファイルを転送するソフトウェア)とTera Tarm(端末エミュレータ)をダウンロードしていない場合はしておきましょう。
- 作成した入力(.gjf or .com)ファイルを転送ソフトで各自に割り当てられた計算サーバーのデレクトリに移動させます。
- ターミナルエミュレータから実行コマンドを入力してジョブを実行します。
- 計算終了後、転送ソフトで出力(.out or .log) ファイルを自身のパソコンに戻し、結果を解析します。
以下に一般的なGaussianの実行コマンドを載せておきます。クラウド利用の場合、各計算機サーバごとに実行方法が用意されていますので、調べてみてください。
Unix/Linax版のGaussian16(or 9)の実行コマンド
cd …/○○○○ # 割り当てられた計算サーバーのデレクトリへのパスへの移動
…/○○○○/g16 inputファイル名.gjf outファイル名.out # 実行コマンド
岡崎共通研究施設計算化学研究センターの実行コマンド
…/○○○○/g16 inputファイル名.gjf # 実行コマンド
分子研の計算機を利用する場合は上のようにoutファイル名を省略することが出来ます。
5. 出力(out)ファイルの確認(安定構造かチェック)
最後に作成された出力(.out or .log) ファイルを確認し、計算が終了してることを確認していきましょう。
確認することは以下の2つです。
- エラーが出ずに正常に終了しているか。
- 振動数計算の結果、虚振動が出ていないか。
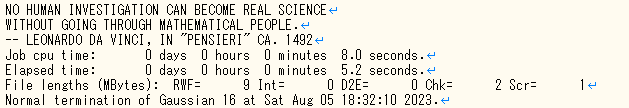
1. は出力(.out or .log) ファイルの最後の行を見れば確認することが出来ます。図のように正常に終了していれば完了です。一方でエラーが出ている場合、エラーが出た理由を確認し、再度入力ファイルを作成して計算する必要があります。
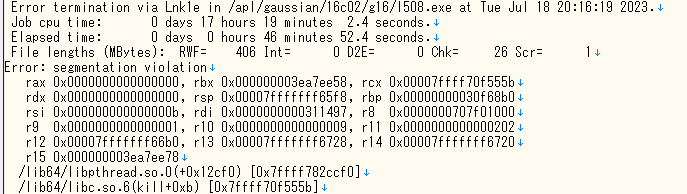
2.は安定構造になっているかを確かめるために行います。振動数計算をしていても、上で述べたように虚振動が出ている場合は、ポテンシャルの谷ではなく、鞍点で収束しているため、安定構造ではありません。しっかりと安定構造で収束しているかを以下の手順で確認してください。
テキストエディタで確認する方法
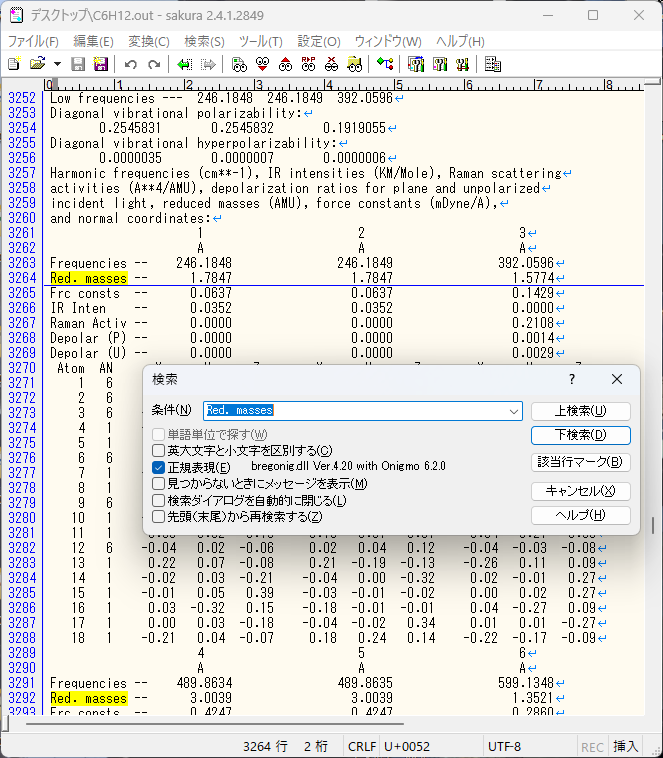
①テキストエディタで出力(.out or .log)ファイルを開いて”Red. masses”の文字列を検索してください。(”ctrl+F”で検索コマンドが表示されます)
②一番初めに出るRed. massesの上の行のA列(今回は246.1848)が正の値を取ってれば安定構造になっています。
また、振動数計算を行っておけば、各構造のゼロ点エネルギーやエントロピー、ギブズエネルギー等が計算される為、安定構造同士でエネルギーを比較することが出来ます。そのため、構造異性体の中から最安定構造を得ることや、遷移状態とのギブズエネルギーの差を出すこともできます。(検索する場合はSum of electronic and thermal Free Energiesを出力ファイル内で計算してください。)
最後に
いかがでしょうか!Gaussianは簡単に構造最適化を計算できることが特徴ですが、特に入力ファイルを正しく作成しないと、単純なエラーに引っ掛かったり、無意味な計算をしてコストが無駄なコストが掛かることもよくあります。時々この記事を見直して、効率的な量子化学計算ライフを送ってください!
おまけ
構造最適化のテクニック エラーを回避するための入力ファイル最終チェックリスト
- 入力ファイルの最後に空行はあるか?
- 各セクションの区切り空行は入っているか?
- 全角文字が入っていないか
- 使用するメモリ量・プロセッサ数は適切か
- Gaussianキーワードは正しいスペルか?(汎関数やopt, freqなど)
- チェックポイントファイル(.chk)の名前に特殊文字([,][.][[\][:][¥]など)が入っていないか
- 電荷とスピン多重度は正確か?
- 分子構造指定は適切か?
参考
[1] 平尾公彦、武次徹也「新版 すぐできる 量子化学計算ビギナーズマニュアル」P. 33-51 [2] 西長亨、本田康 「有機化学者のための量子化学計算入門 Gaussianの基本と有効利用のヒント」 第3・4・5章 P. 32-91
Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

