
この記事は、Avogadroを使用して分子振動やIRスペクトル、QTAIM法による分子間相互作用を描写する方法についてのわかりやすく解説します。記事では、
Avogadroのダウンロードから、解析に必要なファイルの作成方法、解析結果を描写し加工するところまで解説しています。
この記事を学ぶことで、Gaussviewで行うよりも簡単に簡単に分子の振動や相互作用を描写できるようになり、また発表資料の作成に役立つスキルを習得できるので是非最後まで読んでみてください!
今回使用するGaussianおよびAvogadroは以下の目的で利用しています。
Avogadro : IRスペクトルの可視化、振動の可視化、分子間相互作用の可視化
Gaussian : 量子化学計算の実行、WFNファイルの出力
動作環境:Windows 11 version 22H2、Gaussian16、Avogadro 1.2.0
Windows 11(22H2), Gaussian16、Avogadro 1.2.0

Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!
Avogadroとは?

Avogadroは、分子の3D構造をモデリングし、分子座標を作成および視覚化するための無料で利用できるソフトウェアです。主な特徴は以下の通りです:
- 分子モデリング: Avogadroは、分子の原子や結合を直感的に配置し、3Dモデルの分子座標を構築するのに役立ちます。また、小さな分子だけでなく、タンパク質や結晶構造などの大きな系もモデリングすることができます。
- 多くのフォーマットのサポート: Avogadroは、多くの分子ファイルフォーマットをサポートしており、他の化学ソフトウェア(Gaussian やGAMESS等)とデータを簡単に共有できます。これにより、異なるソフトウェア間でデータを円滑にやり取りできます。
- オープンソース: Avogadroはオープンソースソフトウェアで、無料で利用できます。また、コミュニティによって開発され、継続的に改良が行われています。
- プラグインサポート: Avogadroはプラグインアーキテクチャを採用しており、さまざまな追加機能を統合できます。これにより、ユーザーは必要に応じてソフトウェアの機能を拡張できます。
総括すると、Avogadroは化学者や研究者にとって非常に役立つツールであり、分子のモデリングと計算に関する多くの機能を提供しています。特に高価なGaussviewと異なり、無料で利用できることが大きな特徴です。
また、Gaussviewでは分子画像がPNGファイルで出力できないのに対し、AvogadroはPNGファイルでの出力に対応しているため、セミナーや学会の資料作成にも有用です!
今回はこのAvogadroを用いて簡単に分子振動・IRスペクトルを可視化し、さらにQTAIM解析を行っていきます!
Avogadroのインストール
早速、Avogadroをインストールしていきましょう!
Avogadroは以下の公式サイトでダウンロードすることが出来ます。
Avogadroのダウンロードページを開くと以下のようなページに遷移するので、タブ内中央の”Download”をクリックしてください。自動的にダウンロードが開始されます。
次に、ダウンロードした”Avogadro-1.2.0n-win32.exe”ファイルを開いてください。ここの1.2.0の部分はAvogadroのバージョンを示している為、変わる可能性があります。開くと管理者権限を問われる画面になりますので、許可してください。
許可するとセットアップ画面が現れます。以下の手順に従ってセットアップを進めてください。
①下の次へ(N)を押してください。
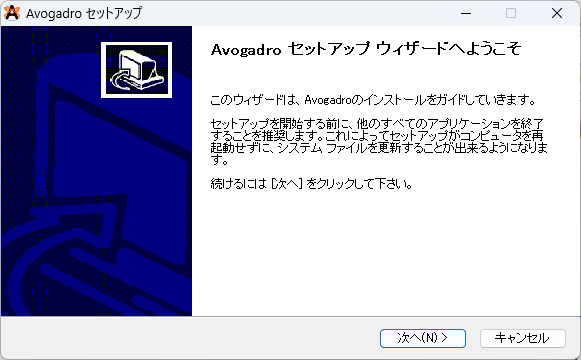
②ライセンス契約書に同意します。
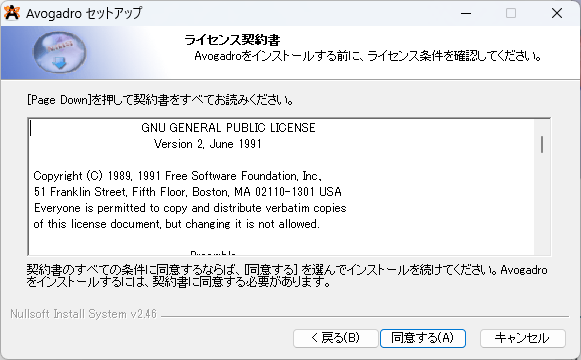
③インストールオプションを選択します。ここではAvogadroのPathを自動でPCに設定してくれます。画像のように選択してください。ログインしているユーザーアカウントのみでPathを設定する場合は、3つ目を選択しましょう。また、すぐ使えるようにCreate Avogadro Desktop icon を選択しておきましょう。
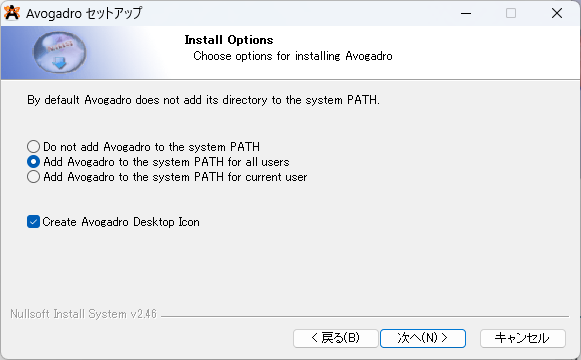
④最後にインストールするフォルダを選択してください。選択後、次へ(N)を押すとインストールが始まります。
分子振動の表示
AvogadroではGaussianを使って振動数計算を行った結果から得られる分子の振動を可視化することが出来ます。
振動数計算は構造最適化計算と組み合わせて行います。今回はベンゼンを対象に解説していきます。Gaussianによる構造最適化+振動数計算の入力ファイルは以下のように作成して計算を実行してください。詳しい入力ファイル作成方法はこの記事に記載しています。是非参考にしてください。
ベンゼンの構造最適化+振動数計算を行う際の入力ファイル
%Chk=benzen.chk
#p B3LYP/6-31G(d) Opt freq pop=npa
benzen
0 1
C -3.15080 2.57109 -0.00000
C -4.24025 1.69336 -0.00000
C -4.02484 0.31100 0.00000
C -2.71998 -0.19363 0.00000
C -1.63052 0.68410 -0.00000
C -1.84593 2.06646 -0.00000
H -3.31744 3.64052 -0.00000
H -5.24973 2.08376 -0.00000
H -0.62104 0.29370 0.00000
H -1.00310 2.74550 -0.00000
H -4.86768 -0.36804 0.00000
H -2.55333 -1.26306 0.00000
計算によって得られた出力ファイル(.outまたは.logファイル)をAvogadroで開くと、「Vibrations」のツールバーが自動的に開きます。
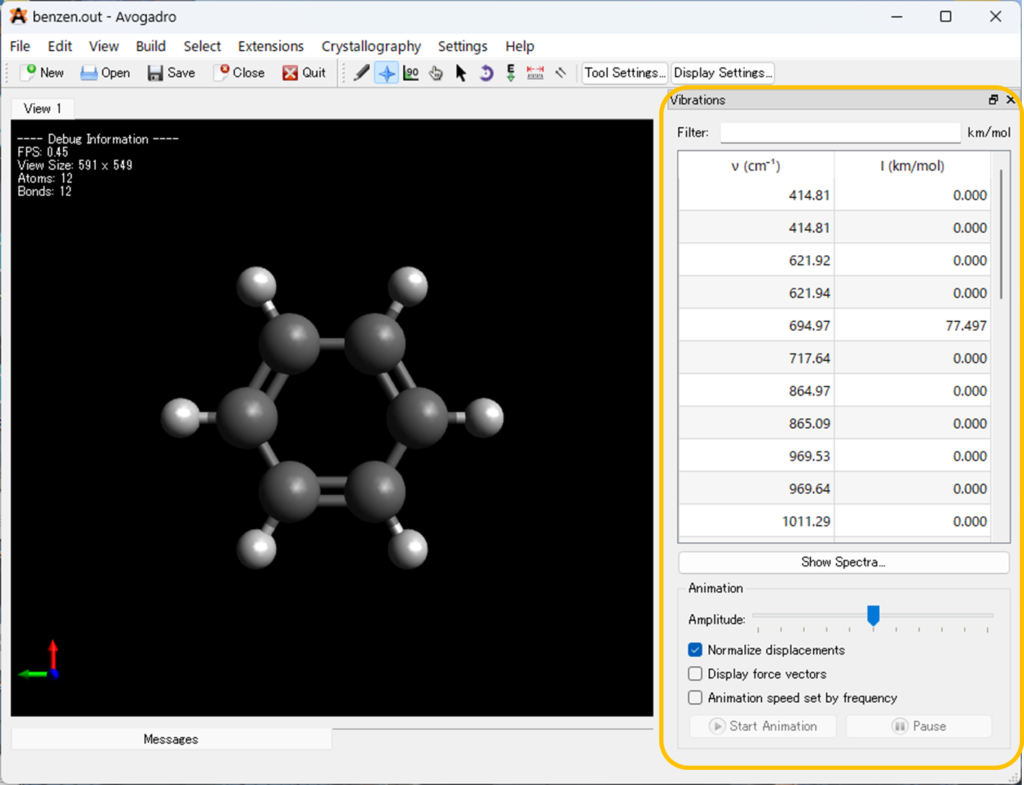
「Vibrations」のツールバーを使用すると、分子のさまざまな振動を表示できます。①で任意の振動数を選択し、②の「Start Animetion」をクリックすると振動が始まります。
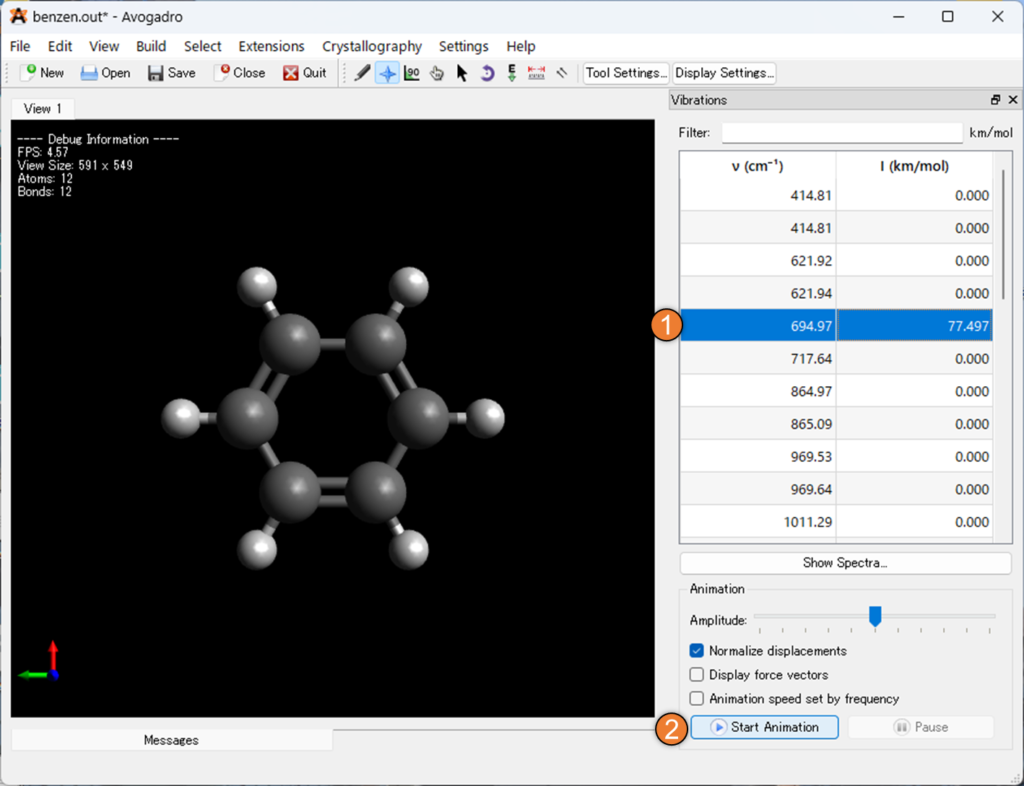
以下の画像の①「Amplitude」は振動の大きさを編集することが出来る機能です。
また、②の「Display force vector」を選択することで、存在する原子の力ベクトルを表示し、③の「Animetion speed set by frequencey」を選択することで振動数によってアニメーション速度を調整することができます。
IRスペクトルの表示
次にIRスペクトルの表示方法について説明します。
Gaussian からの出力ファイルを使用するとラマンスペクトルなど他のタイプのスペクトルも表示可能です。
こちらも先ほどと同様に、構造最適化+振動数計算を実行して得られた出力ファイルを開くと、「Vibrations」ツールバーが自動的に開きます。
スペクトルを表示
「Vibrations」の ツールバーの [Show spectra …] をクリックするか、[Extension] メニューで [spectrum…] を選択すると、分子のIRスペクトルが開きます (下に表示)。
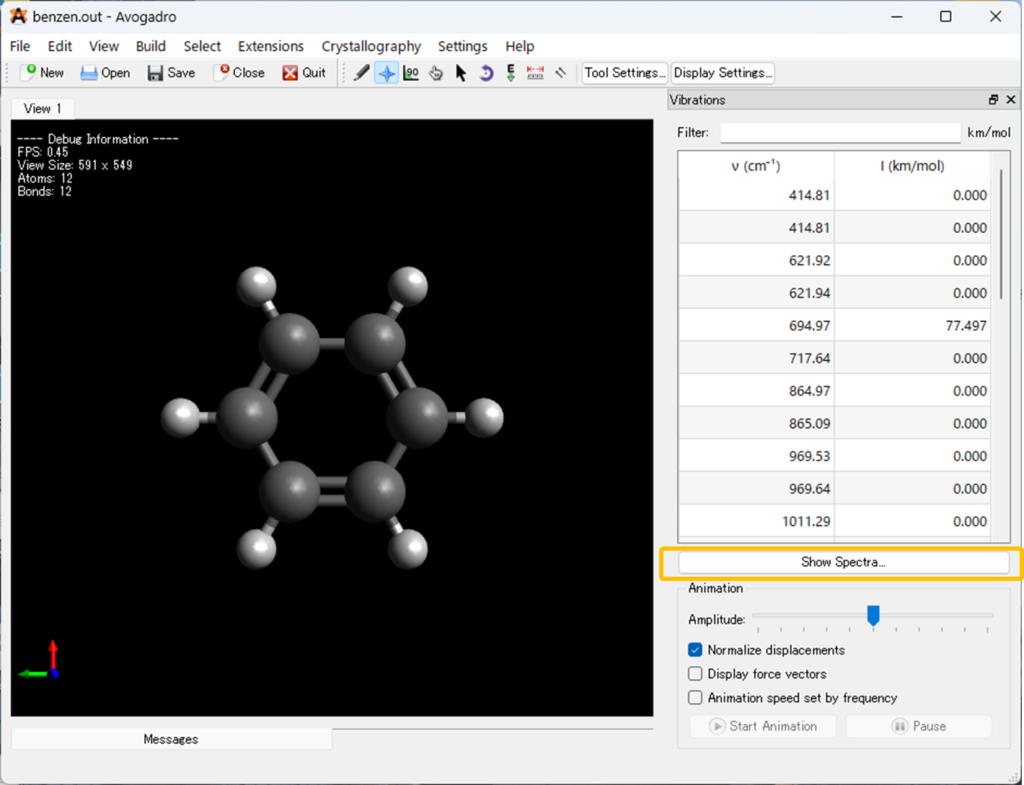
以下のようなIRスペクトルが表示されると思います。ここで①の「Advanced»」を選択すると、必要に応じてスペクトルの編集をAvogadro上で行うことが出来ます。
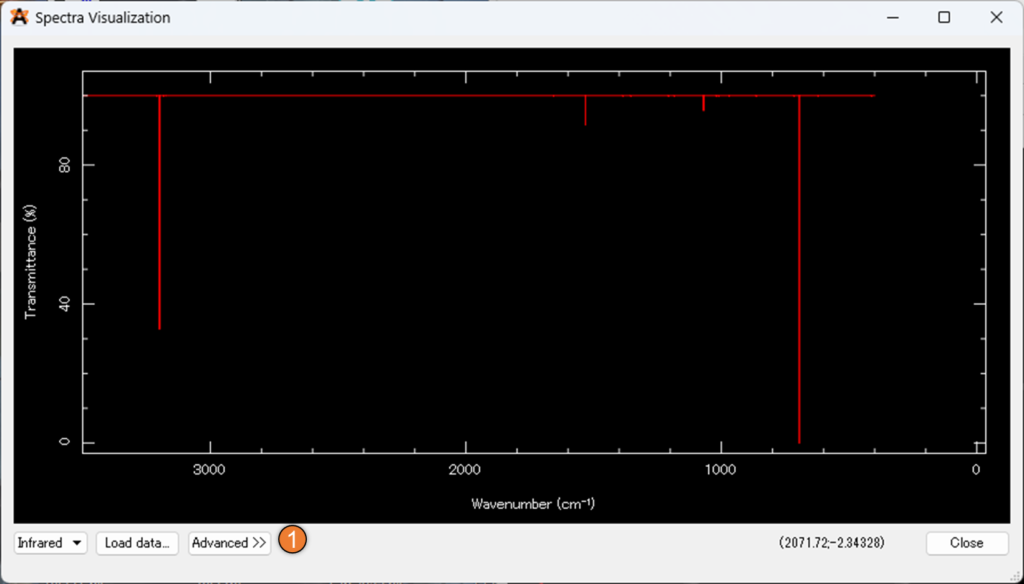
たとえば、以下の画像の②のLightを押すことで背景を黒から白に変更したりできます 。
設定後は③の「infrared Spectra Settings」を選択してください。
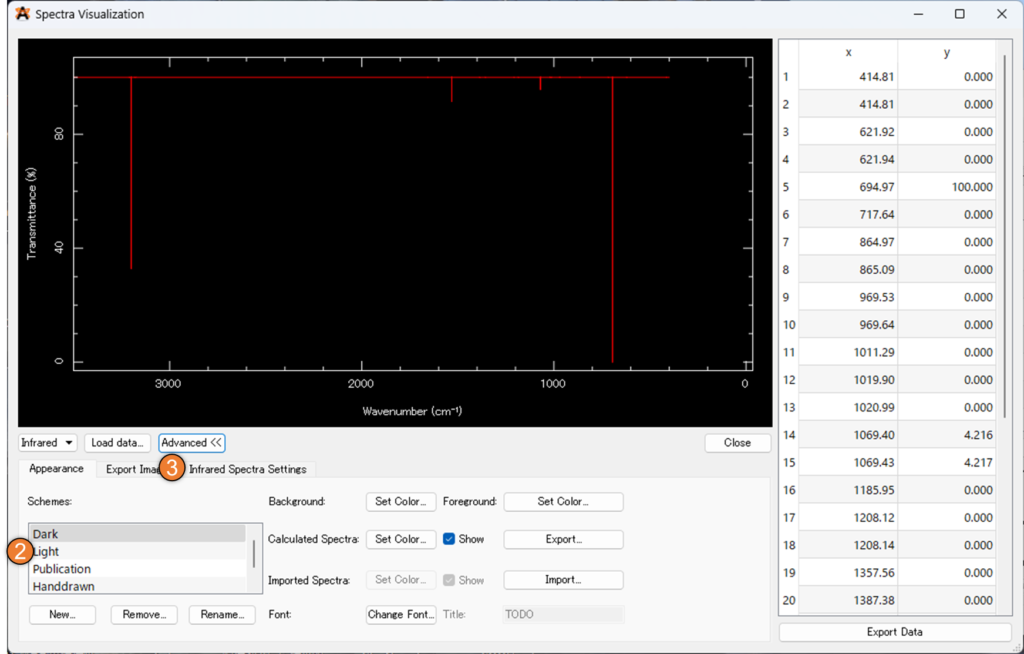
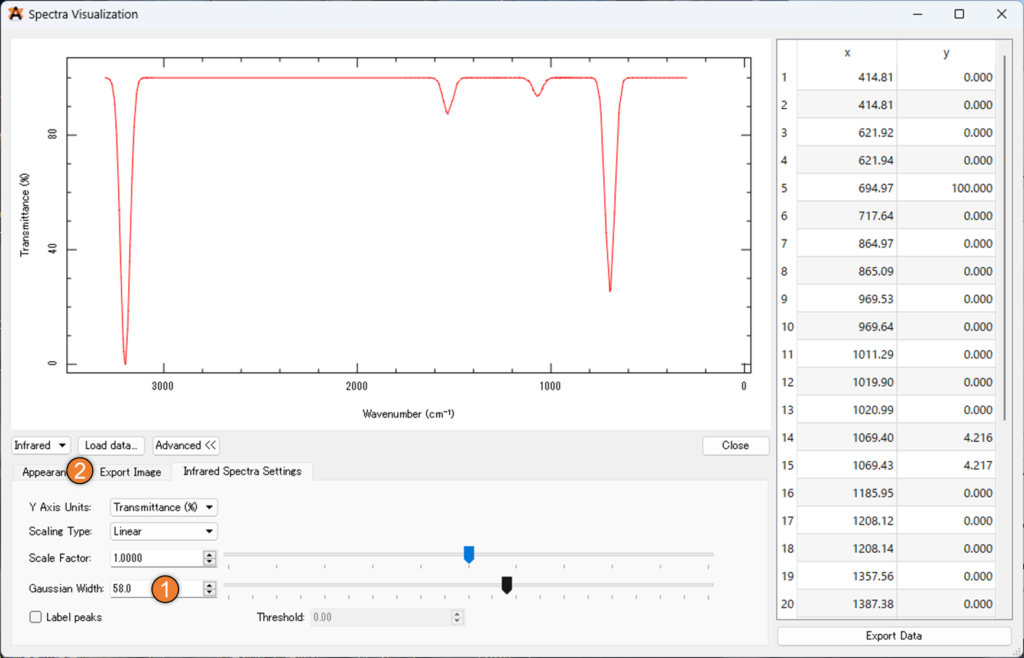
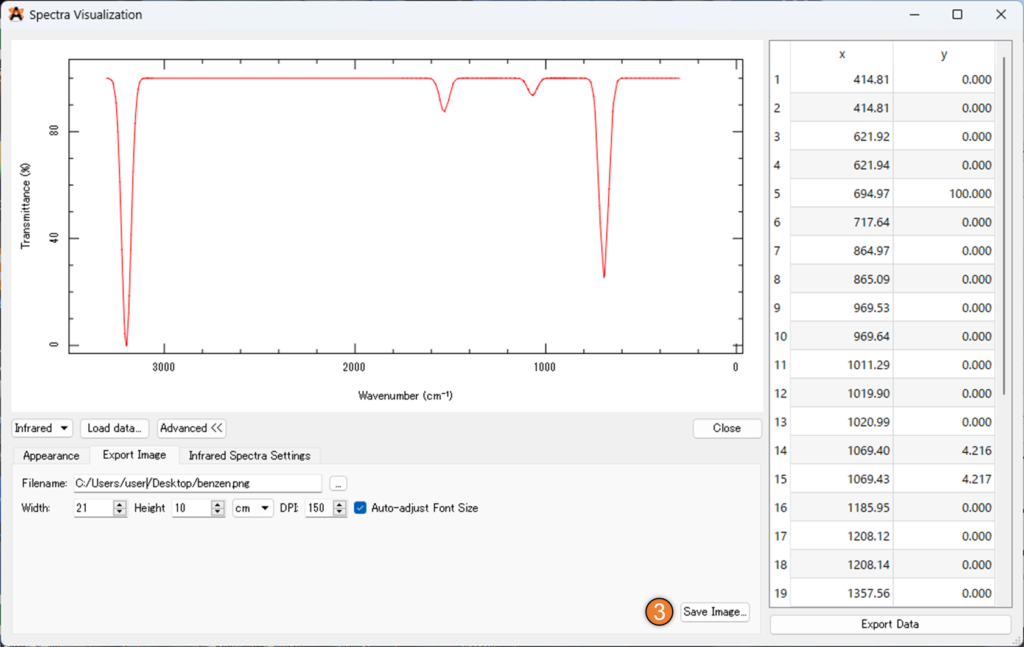
「infrared Spectra Settings」では、% 透過率を % 吸光度に変更することが出来ます。また、下の画像①の「Gaussian Width」を大きくするとピーク位置に指定の幅のガウス関数を置いてくれるため、実験結果と類似した理論スペクトルを表示させることが出来ます。
最後に②の「Export Image」からファイル名を設定し、③の「Save Image..」を選択することで、スペクトルの画像を得ることが出来ます。
分子間相互作用の可視化(QTAIM解析)
次にAvogadroで解析できる、QTAIM解析について紹介します。
QTAIM解析とは
QTAIM (The Quantum Theory of Atoms-In-Molecules) 法は、電子密度を分子内で詳細に調査する方法で、化学結合と相互作用の性質の分析、評価、分類することが出来ます。この方法では、電子密度の局所極大点や局所極小点を見つけ出し、その点における電子分布や結合特性を評価します。つまり、QTAIM法は分子の中で電子がどこに集まり、どこから影響を受けているかを探し、それが分子内での化学結合や反応にどのように影響を与えているかを調査します[2]。
Avogadro には、Richard Bader 教授と彼のグループが開発した QTAIM 分析のサポートが含まれるようになりました。この技術により、Avogadroを用いると量子力学的電子密度から直接結合結合と結合次数を決定することができます。
WFNファイルの作り方
Avogadroでは.wfnファイルを用いることで、QTAIM 拡張機能を使用することが出来ます。Gaussianをはじめとする多くの量子化学ソフトウェアでは、計算の出力から WFN 形式のファイルを作成できます。以下ではGaussianを用いて.wfnファイルを作成する方法を解説します。
- 新規の構造最適化計算で.wfnを生成する場合
新しい計算では、キーワードOutput=wfnを指定し、座標の後にfilename.wfnを指定します。
また通常、Windows版のGaussianなどで生成すると.chkや.wfnはC:\g16w\scriptなどのGaussianアプリが収納されているフォルダに生成されている場合があります。
目的のフォルダに.chkや.wfnを生成したい場合は、C:\Users\user\Desktop\B2H6.wfn
のように絶対パスを設定することをお勧めします!
Gaussianを使って.chk ファイルと .wfn ファイルの両方を生成するB2H6の構造最適化入力ファイル
%chk=C:\Users\user\Desktop\B2H6.chk
#n B3LYP/3-21G Opt output=wfn
Title Card Required
0 1
B -2.65538534 0.44370962 -0.29611581
B 0.38336988 0.80483249 -0.12380108
H -1.23848846 1.49043622 -0.05646593
H -1.02407837 -0.25379666 -0.18215095
H 0.89547746 1.55628824 0.62816972
H 1.07443466 0.10046568 0.52326683
H 0.88796670 0.94307335 -1.18147377
H -3.28342818 0.29532190 0.69178261
H -3.02922973 -0.33900436 -1.09611531
H -3.22064810 1.21819021 -0.98390781
C:\Users\user\Desktop\B2H6.wfn
入力ファイルのそれぞれの説明はこちらの記事を参考にしてください。
- すでに構造最適化計算が実行され、.chkファイルがある場合
すでに実行された計算(.chk ファイルが利用可能)からでも.wfnファイルを生成することが出来ます。
まず、計算が終了した .chk ファイルを指定します。
次に、geom=allcheck オプションを追加すると、すでに収束された構造最適化計算の .chk から安定構造が読みだされ、その構造を基に .wfn ファイルが出力されます。そのため、今回の入力ファイルでは分子指定セクションは省略します。
最後に .wfn ファイルのファイル名を指定して、準備完了です。そのあとの空行にも意味がありますので、以下の入力ファイルを参考にしてください。
Gaussianを使って*.chk ファイルから.wfn ファイルを生成する入力ファイル
%chk=C:\Users\user\Desktop\B2H6.chk
#n output=wfn B3LYP/3-21G geom=allcheck guess=only
C:\Users\user\Desktop\B2H6.wfn
孤立電子対・電荷シフト結合・分子間相互作用を描写
では、AvogadroでQTAIM解析を行っていきます。
[Extension] メニューで [QTAIM] を選択し、 [Molecular Graph with Lone Pairs] を選択すると.wfnファイルの選択画面になります。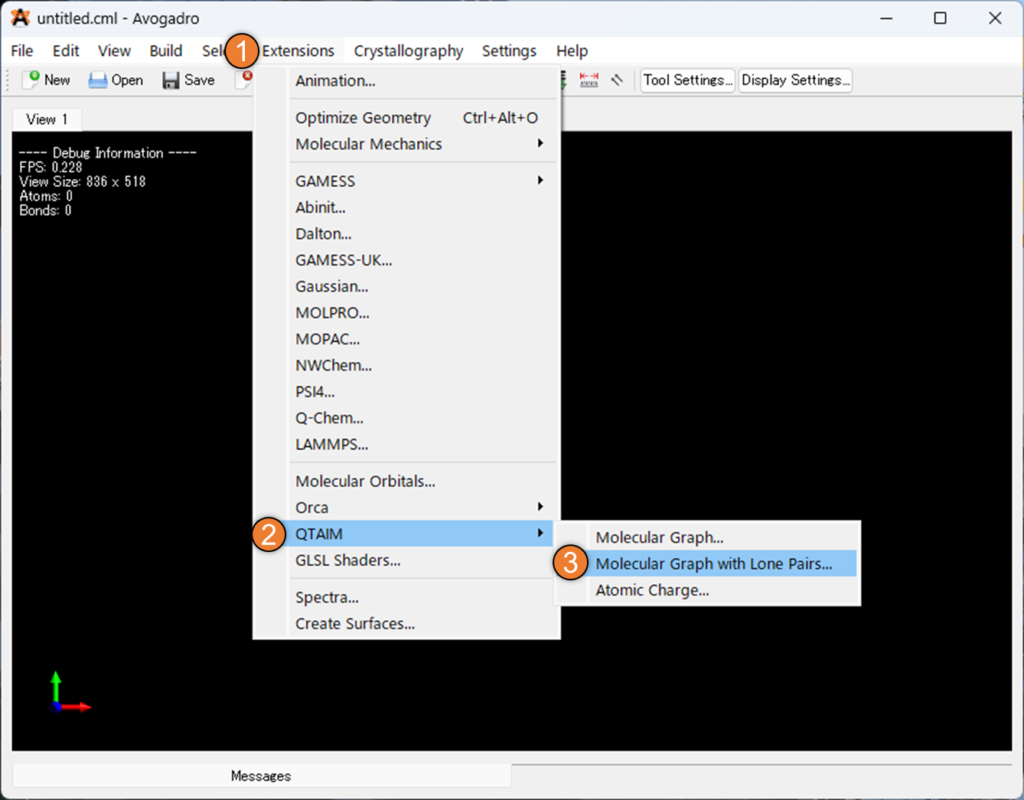
.wfnファイルの選択画面で対象の .wfn ファイル (今回はB2H6.wfn) を選択すると以下のように解析が始まります。
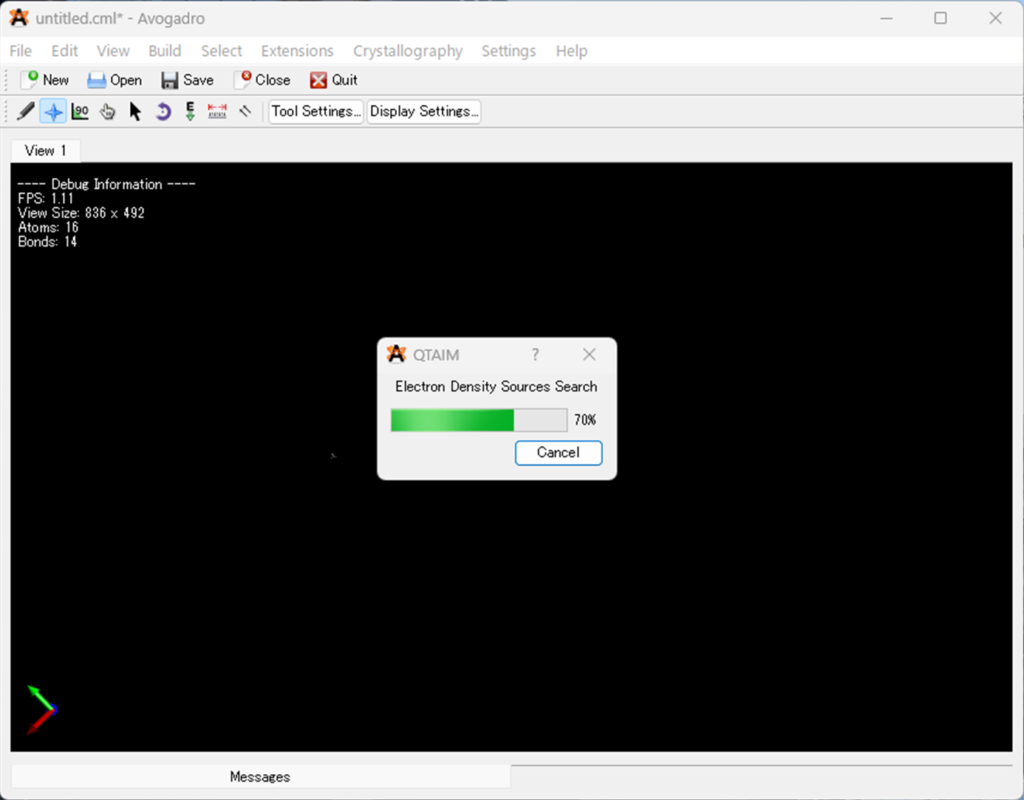
Avogadro の QTAIM サポートは原子を読み込み、AIM 解析を使用して結合と結合次数を決定し、さらに解析を続けます。
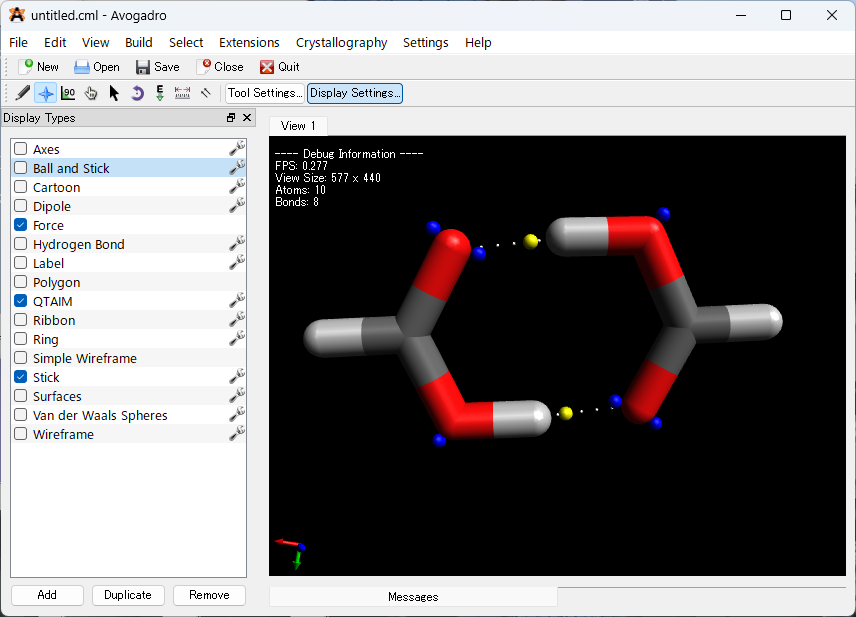
解析後、最初は何も変わっていないように見えるかもしれません。Avogadro には、QTAIM 分析の結果を表示するための別の表示タイプが含まれています。以下の画像①のように「Display Settings…」を選択後、②のQTAIMを選択してください。
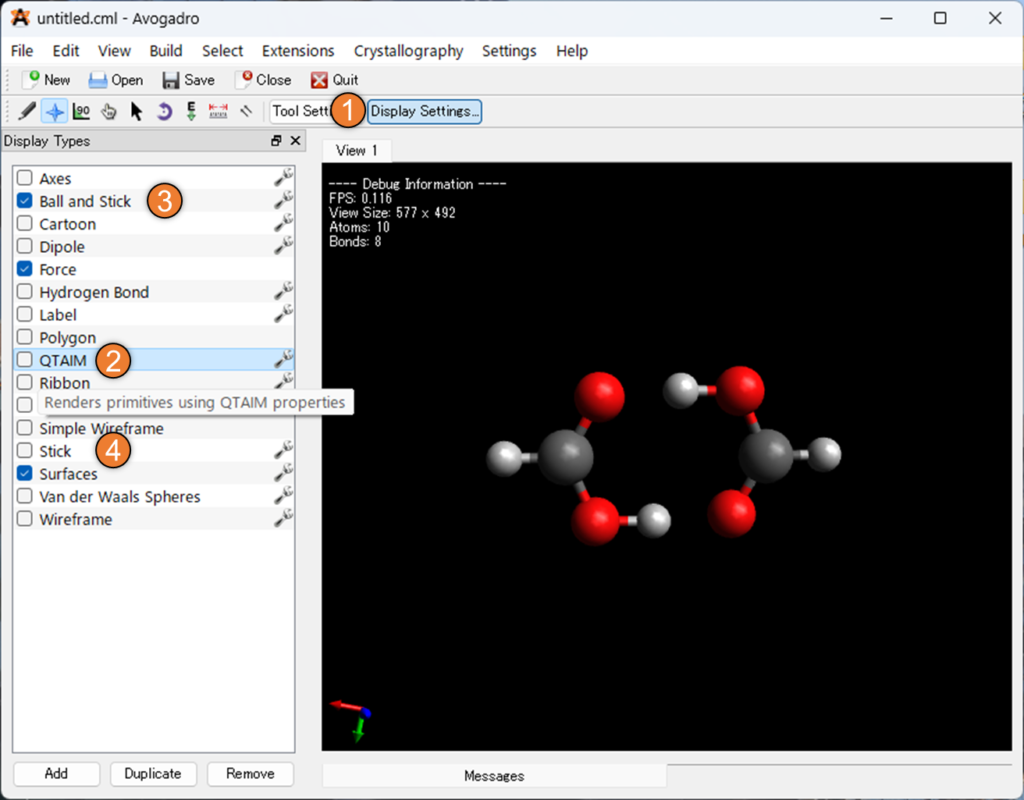
ここでは、孤立電子対、結合臨界点などが表示される QTAIM 表示を有効にしました。上の画像③・④のように、「Ball and Stick」を外し、「Stick」にチェックを入れることで、孤立電子対や水素結合と考えられる弱い結合を分かりやすく描写することができました。
すべての QTAIM の結果を表示するには、「Stick」及び「Ball and Stick」をオフにすることで表示することが出来ます。ここでは、「Stick」及び「Ball and Stick」表示タイプもオフにして、B2H6の QTAIM ビューのみが残ります。
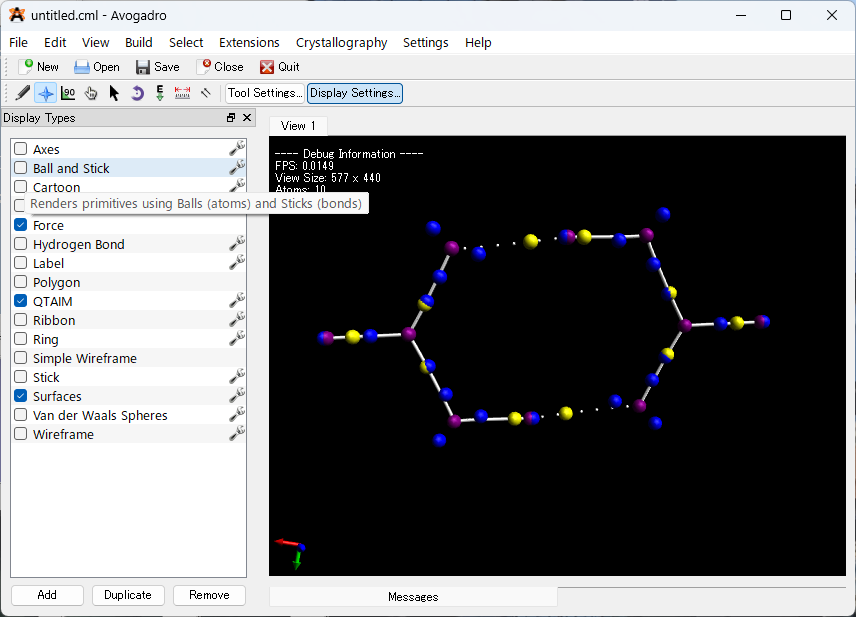
上の画像は見慣れない画像ですが、それぞれ以下のような特徴を示しています。
- 小さな白い球の結合パス: 電荷シフト結合をはじめとする弱い結合(結合臨界点のラプラシアン > 0.0の 場合)
- 結合パス: 共有結合などの強い結合(白い円柱の場合 ラプラシアン<0)
- 核臨界点: 紫色の球体(今回はC,O,H原子)
- 結合臨界点: 黄色の球体
- 電子密度源 (非共有電子対など): 青い球体
以上のようにQTAIM解析は、電荷シフト結合を含むさまざまな種類の結合を理解し、結合の性質を詳細に解析するのに役立ちます。
最後に
最後まで読んでいただき、ありがとうございました!いかがでしょうか。特に、QTAIMは計算化学の分野で近年見られるようになってきた解析手法であり、Avogadroを用いれば簡単に解析できます。気軽に活用してみてください!
参考
[1] Avogadro 公式 https://avogadro.cc/ (参照2023-11-05) [2] P. Julien et. al., QTAIM Analysis in the Context of Quasirelativistic Quantum Calculations, J. Chem. Theory Comput.10, 11, 4830 (2014) [3] Chemistry Stack Exchange https://chemistry.stackexchange.com/questions/123984/how-can-i-convert-gjf-or-chk-file-to-wfn-file (参照2023-11-05)
Gaussianを使った量子化学計算の初心者向け技術書を販売中
¥2,000 → ¥1,000 今なら50%OFF!!
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

