
scRNA-seqデータ解析である程度の可視化はできたけど、複数のサンプルを比較して解析する場合がわからないかと思います。例えばコントロール群と刺激群を統合して同じようにプロットしたくなってきます。
この記事ではscRNA-seqデータのコントロールと刺激サンプルをバッチエフェクト補正した後に統合して解析する方法を紹介します。
コントロール群と刺激群を比較できるようになるだけで複雑な細胞タイプに対する比較分析が可能になります。挑戦してみましょう。
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB seurat 4.3.0

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
scRNA-seqデータの統合について
今回はseuratのハンズオンの中にあるこちらの記事について解説していこうと思います。
scRNA-seqデータの統合
2つ以上のシングルセルデータセットを統合して分析する方法がSeurat によって提供されています。
例えば、以下の図のようにデータ統合を行うことで、コントロール(CTRL)と刺激(STIM)がほぼ一致するようなUMAPが描かれていることが分かるかと思います。
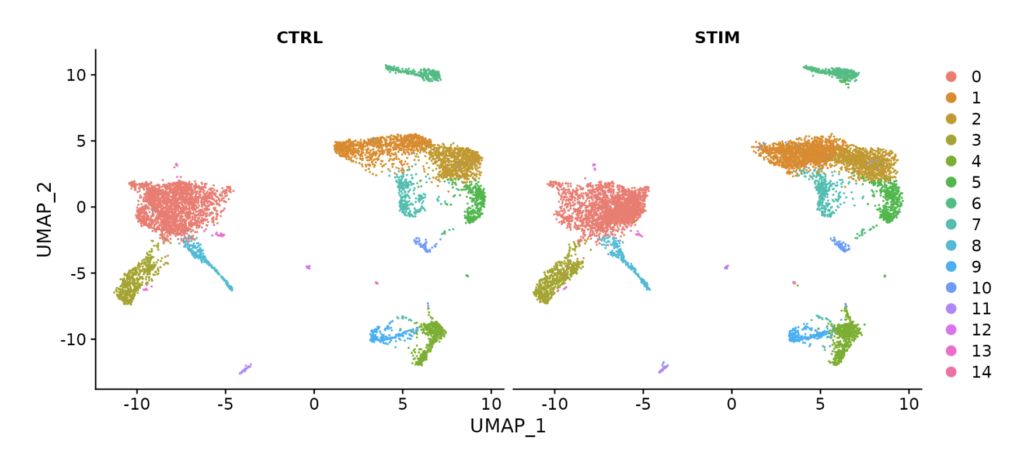
この統合されたサンプルを使えば、各遺伝子でコントロールに比べ発現が上昇した遺伝子(下図は、IFI6の例)を確認することができます。
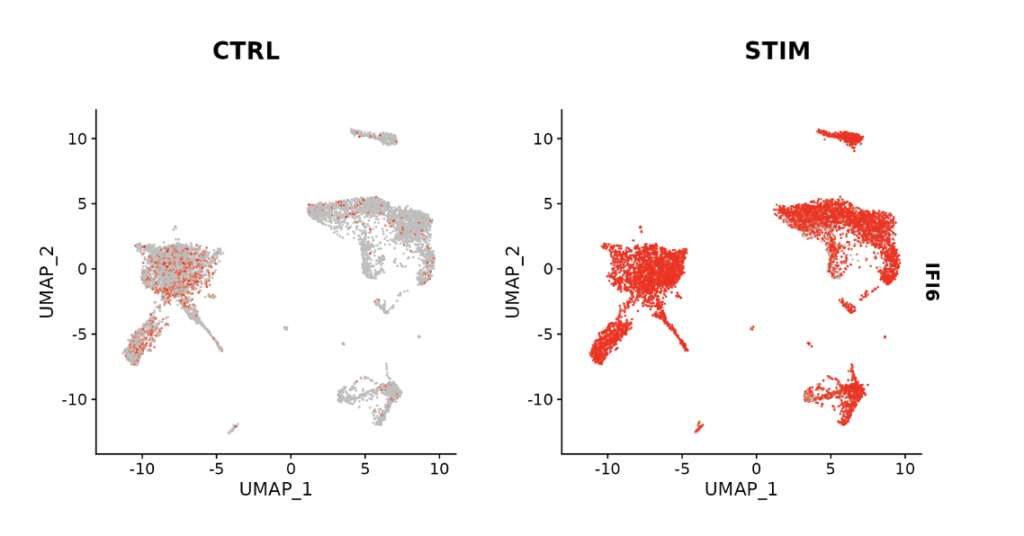
https://satijalab.org/seurat/articles/integration_introduction より一部改変して引用
アンカーとは?
アンカーとは、異なるシングルセルRNAシーケンス(scRNA-seq)データセット間で生物学的に一致している(または非常に類似している)細胞ペアを見つけるためのフレームワークの一部です。データセット間で一貫して変動する遺伝子(フィーチャー)を用います。このような細胞ペアは、複数の状態や時間点、または実験間で一致している特定の生物学的特性(例えば、特定の細胞型や発現パターン)を持っています。アンカーには以下のような役割があります。
- バッチエフェクトの補正: 異なる条件や実験からのデータセットを統合する際には、技術的な違い(いわゆるバッチ効果)が生じる可能性があります。アンカーを用いることで、これらのバッチエフェクトを認識し補正することが可能になります。
- 相対的な解釈: 異なるデータセットや条件間での相対的な遺伝子発現の変化(例えば、刺激前後での変化)を評価する際にもアンカーが有用です。
- 生物学的インサイト: アンカーを正確に識別できれば、細胞群がどのように遷移するのか、どの細胞が互いに関連しているのか、といった生物学的な洞察が得られます。
scRNA-seqデータセット間のバッチエフェクト補正と統合
それではscRNA-seqデータをバッチエフェクト補正後に統合する実装方法を紹介していきます。
まずはseuratオブジェクトを作成していきます。今回使うデータセットはSeuratData パッケージを使用して”ifnb” と呼ばれるデータセットをインストールしていきます。
library(Seurat)
library(SeuratData)
library(patchwork)
# install dataset
InstallData("ifnb")
# load dataset
LoadData("ifnb")ifnbデータセットの中身にはIMMUNE_CTRLとIMMUNE_STIM の2つの実験ラン(Run)が格納されています。ちなみにorig.identカラムは、各細胞の元のサンプルやランを示すために使用されます。

このコードは、各刺激状態ごとにデータを正規化し、変数遺伝子を特定することで、後の分析(例:クラスタリング、遺伝子発現の比較など)でより精度の高い結果を得られるように前処理を行っています。さらに、複数のデータセットを効果的に統合するための準備も行っています。
# split the dataset into a list of two seurat objects (stim and CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
# normalize and identify variable features for each dataset independently
ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {
x <- NormalizeData(x)
x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)
})
# select features that are repeatedly variable across datasets for integration
features <- SelectIntegrationFeatures(object.list = ifnb.list)詳しいコードの解説は以下です。
SplitObject(ifnb, split.by = “stim”): ifnbというSeuratオブジェクト(これが単一細胞RNA-seqデータを含む)をmeta.dataの中にある”stim” 列(刺激状態を表すメタデータ)に基づいて2つの異なるSeuratオブジェクトに分割します。これにより、刺激状態ごとにデータを個別に分析できるようになります。

lapply(...): lapply関数を使用して、各Seuratオブジェクト(刺激状態ごとのデータ)に対して次の操作を行います。
NormalizeData(x): データを正規化します。これは、各細胞のRNAの総量でカウントをスケーリングすることにより行われます。
FindVariableFeatures(...): 変数遺伝子(各細胞集団で異なる発現を示す遺伝子)を探します。このステップは、後のクラスタリングや他の解析で重要な遺伝子を特定するために重要です。
SelectIntegrationFeatures(object.list = ifnb.list): 2つ以上のデータセットを統合する前に、それらのデータセット間で一貫して変動する遺伝子(フィーチャー)を選択します。このステップは、後でFindIntegrationAnchors()やIntegrateData()のような関数を使用してデータセットを統合する際に重要です。
ちなみに最初に一回seuratオブジェクトを分割するのはなぜ?と思う方もいらっしゃるかもしれません。以下のような理由でオブジェクトを一度分割します。
- 個々のデータセットの特性を理解する: それぞれの状態(この場合は刺激状態か非刺激状態)に対する細胞の反応は異なる可能性があります。この違いを理解するためには、それぞれの状態を独立して解析する必要があります。この独立した解析によって、各状態に特有の変数遺伝子や細胞クラスターを特定できます。
- バッチ効果の補正: それぞれの状態に対して独立した解析を行うことで、後の統合フェーズでバッチ効果(技術的な変動)をより効果的に補正できます。これは特に、各状態が異なる実験設定で得られた場合に重要です。
- 高度な比較解析: 独立したオブジェクトとして各状態を分析することで、状態間での遺伝子発現の違いや細胞クラスターの違いなど、より高度な比較解析が可能になります。
- 統合の精度: Seuratの統合メソッドは、各データセット(この場合、各状態)から選ばれた変数遺伝子を用いて「アンカー」(相当する細胞状態を示すポイント)を見つけ、それを基にデータセットを統合します。このプロセスには、各データセットが独立して前処理された情報が必要です。
以上のような理由から、多くのケースで最初にデータを独立したオブジェクトに分割し、それぞれに対して前処理と基本解析を行った後で統合するというアプローチが取られます。
次にアンカーを使って2つのデータセットを統合をしていきます。こちらのコードは複数のscRNA-seqデータセットを統合し、バッチ効果を除去するコードです。
# Find integration ancher
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, anchor.features = features)
# this command creates an 'integrated' data assay
immune.combined <- IntegrateData(anchorset = immune.anchors)FindIntegrationAnchors関数: この関数は、指定された特徴量(変数遺伝子)に基づいて、複数のscRNA-seqデータセット間で「アンカー」と呼ばれる一致する細胞ペアを見つけます。object.list = ifnb.list: この引数には、統合したいSeuratオブジェクトのリスト(ここではifnb.list)が渡されます。anchor.features = features: 統合に使用する特徴量(遺伝子)のリスト。これは通常、複数のデータセットで変動が大きいと判断された遺伝子です。
IntegrateData関数: この関数は、前のステップで特定されたアンカーを使用して、複数のデータセットを1つの「統合された」データセットに統合します。anchorset = immune.anchors: この引数には、前のステップで特定されたアンカーが含まれています。

ifnbデータセットの場合、2つの条件が同じSeuratオブジェクト(IMMUNE_CTRL とIMMUNE_STIM )に存在していますが、それだけでは統合は行われていません。統合を行ったimmune.combinedには、新たに「integrated」アッセイが生成され、統合されたデータが格納されています。
ifnbオブジェクトの段階では「RNA」アッセイのみですが、これは2つのデータセットの条件を統合せずに一つのSeuratオブジェクトとして保存されていると解釈できます。
次にデータセットが統合されたサンプルを下流解析に持っていきます。
まずは下記コードで、DefaultAssayをintegratedにします。このコードは、immune.combinedというSeuratオブジェクトでのデフォルトのアッセイを”integrated”に設定しています。これにより、次に行う解析はこの統合データを対象とすることが指定されています。
DefaultAssay(immune.combined) <- "integrated"DefaultAssay: Seuratオブジェクトにおけるデフォルトのアッセイ(データセットの特定のビュー)を取得または設定するための関数です。immune.combined: これはSeuratオブジェクトで、多くの場合、シングルセルRNA-seqデータの集合や統合データセットを含んでいます。<- "integrated": これにより、immune.combinedオブジェクトのデフォルトのアッセイが”integrated”に設定されます。”integrated”アッセイは、複数のサンプルや条件からのデータを統合した後のデータを指します。これにより、ユーザーは統合データを基にして分析を進めることができます。
下流の処理を流していきます。seuratで行ういつもの前処理なため詳細は割愛しますが、magrittr ライブラリを用いて%>% operatorでパイプとして処理させています。
library(magrittr) # Load the magrittr package for the %>% operator
# Now you can use the pipe operator
immune.combined <- immune.combined %>%
ScaleData(verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30) %>%
FindNeighbors(reduction = "pca", dims = 1:30) %>%
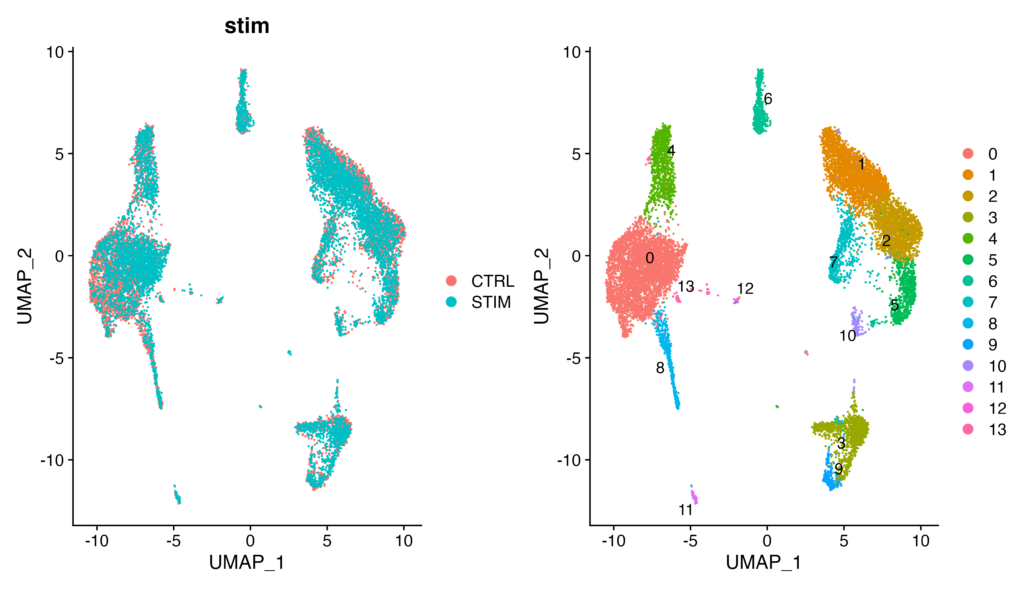
FindClusters(resolution = 0.5)それではUMAPを可視化してみましょう。統合処理のあるなしで比較できるように、immune.combined(統合済みデータセット)とifnb(統合していないデータセット)でそれぞれ出力して比較してみます。
# Visualization
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE, repel = TRUE)
p1 + p2immune.combinedを可視化に用いた場合(統合済みデータセット)
ifnbを可視化に用いた場合(統合していないデータセット)
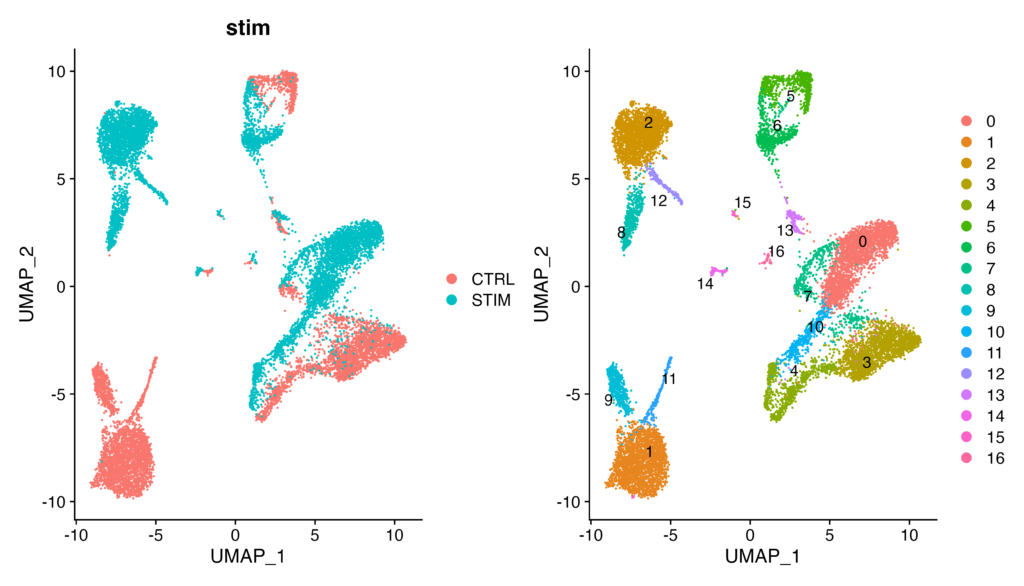
見ての通り、ifnb では、左のUMAPでCTRLとSTIMが全く重なっていないのが確認できるかと思います。そのため、左のクラスタリング結果では、同じ細胞群であろうと予想できるクラスター(例えば0と3は同じ細胞群の可能性があるが、別々にクラスタリングされています)が別のクラスターとして検出されていることが確認でき、解析が不確かになっています。統合処理の重要性がお分かり頂けましたでしょうか。
ちなみに、以下のsplit.by = "stim" を指定するとCTRLとSTIMのグラフを結合できます。
DimPlot(immune.combined, reduction = "umap", split.by = "stim")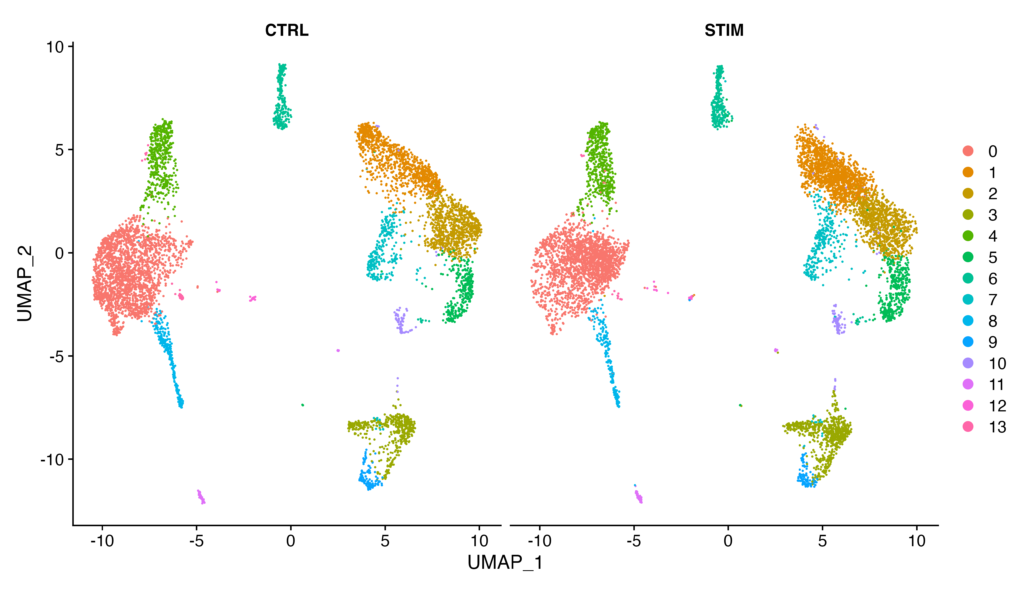
次のセクションに移る前に各クラスターの細胞種を割り振っておきます。(今回は、各細胞種やマーカー遺伝子の特定は割愛します。)
immune.combined <- RenameIdents(immune.combined, `0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T",
`3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "NK", `7` = "T activated", `8` = "DC", `9` = "B Activated",
`10` = "Mk", `11` = "pDC", `12` = "Eryth", `13` = "Mono/Mk Doublets", `14` = "HSPC")
DimPlot(immune.combined, label = TRUE)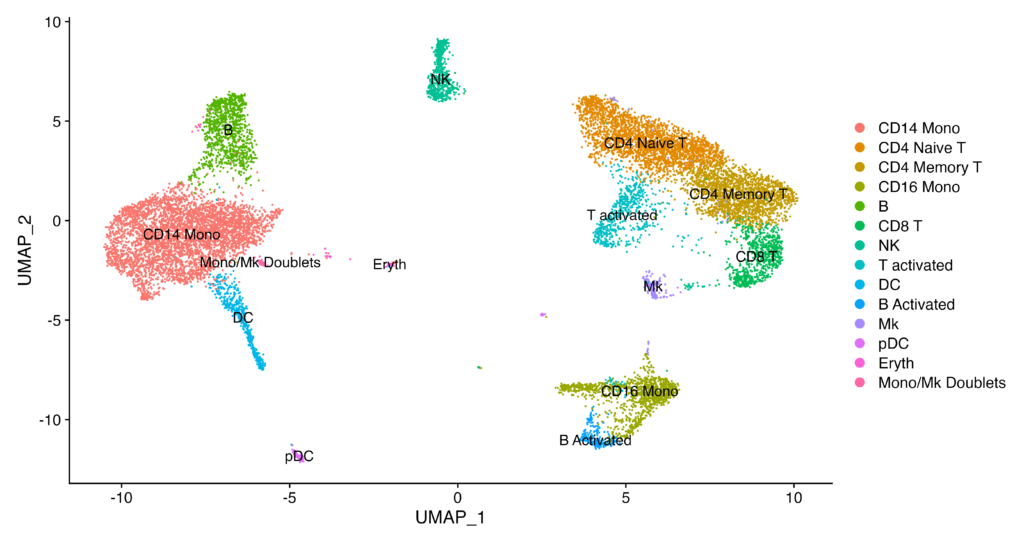
条件間で発現変動遺伝子を特定する
それでは実際にコントロール群と刺激群を比較してみましょう。
このコードは、特定のマーカー遺伝子の発現レベルを、異なるクラスターやカテゴリに基づいて可視化するためのものです。
Idents(immune.combined) <- factor(Idents(immune.combined), levels = c("HSPC", "Mono/Mk Doublets","pDC", "Eryth", "Mk", "DC", "CD14 Mono", "CD16 Mono", "B Activated", "B", "CD8 T", "NK", "T activated","CD4 Naive T", "CD4 Memory T"))
markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5","CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1","GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ", "PRSS57")
DotPlot(immune.combined, features = markers.to.plot, cols = c("blue", "red"), dot.scale = 8, split.by = "stim") +
RotatedAxis()Idents関数は、Seuratオブジェクトから現在のクラスターIDやその他のアイデンティティ情報を取得するために使用されます。この行は、immune.combinedのアイデンティティを再順序化することで、後のプロットなどでの表示順序を制御しています。
markers.to.plot <- c("CD3D", "CREM", …の部分でプロットしたいマーカー遺伝子のリストを定義しています。
Dotplotで以下の出力が出れば成功です。行に各細胞ごとにCTRLとSTIMの比較、列にFeaturesとなる遺伝子が確認できるかと思います。
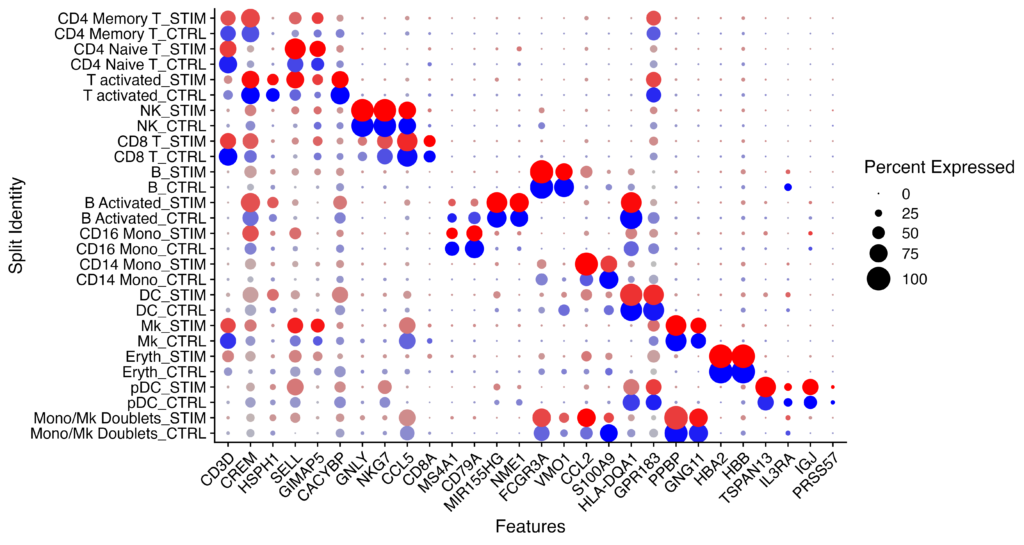
次に条件間で発現が異なる遺伝子を特定していきます。
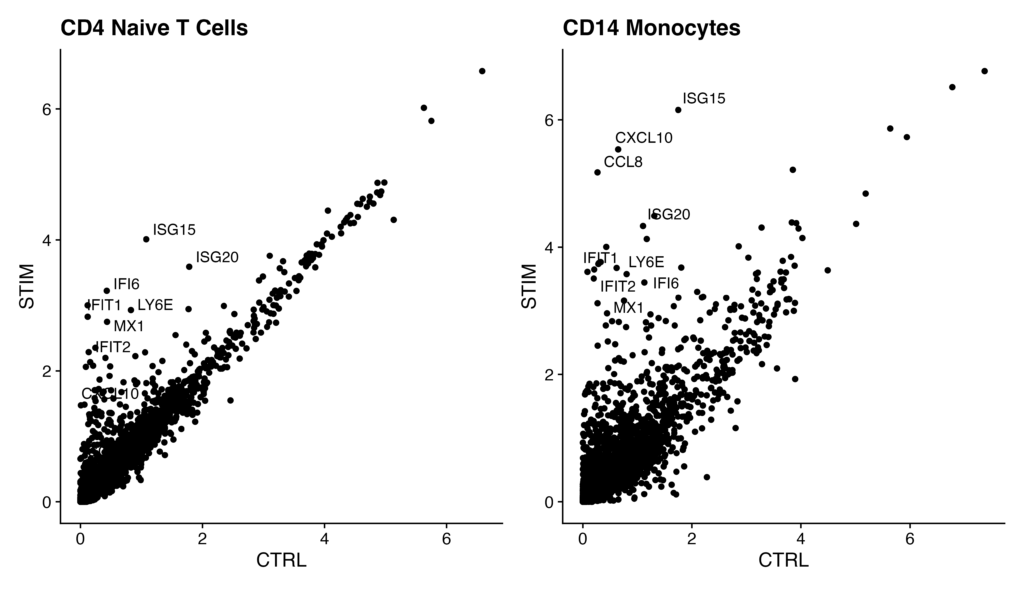
このコードは、刺激された細胞とコントロール細胞の遺伝子発現の違いを視覚的に比較することです。具体的には、ナイーブT細胞とCD14単球集団の発現を比較し、インターフェロン刺激による反応を示す特定の遺伝子をscatter plotで強調しています。
# Load visualization libraries
library(ggplot2)
library(cowplot)
theme_set(theme_cowplot())
# Extract and process data for "CD4 Naive T" cells
t.cells <- subset(immune.combined, idents = "CD4 Naive T")
Idents(t.cells) <- "stim"
avg.t.cells <- as.data.frame(log1p(AverageExpression(t.cells, verbose = FALSE)$RNA))
avg.t.cells$gene <- rownames(avg.t.cells)
# Extract and process data for "CD14 Mono" cells
cd14.mono <- subset(immune.combined, idents = "CD14 Mono")
Idents(cd14.mono) <- "stim"
avg.cd14.mono <- as.data.frame(log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA))
avg.cd14.mono$gene <- rownames(avg.cd14.mono)
# Specify genes to be highlighted in plots
genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8")
# Create scatter plots and label specified genes
p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) + geom_point() + ggtitle("CD4 Naive T Cells")
p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) + geom_point() + ggtitle("CD14 Monocytes")
p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)
# Display plots side by side
p1 + p2コードの処理の流れは以下の通りです。
- 刺激されたナイーブT細胞とコントロールのCD14単球集団の平均遺伝子発現の値を取得します。
- ggplot2とcowplotパッケージを使用して、これらの2つの細胞タイプの遺伝子発現をプロットします。
- 特定の遺伝子(ISG15, LY6Eなど)の点を強調表示してラベルを付けます。
- 最後に、2つのプロット(ナイーブT細胞のものとCD14単球のもの)を並べて表示します。
以下のようなscatterplotが出れば成功です。
以下のコードは、シングルセルRNA-seqデータセット immune.combinedに対して、刺激状態に基づいた細胞タイプの識別、差異発現遺伝子の特定、および特定の遺伝子の発現レベルの視覚化をFeature plotで行っています。
# Create a new identifier combining cell type and stimulation status
immune.combined$celltype.stim <- paste(Idents(immune.combined), immune.combined$stim, sep = "_")
# Backup current cell type identifiers and update to the new combined identifier
immune.combined$celltype <- Idents(immune.combined)
Idents(immune.combined) <- "celltype.stim"
# Identify genes differentially expressed between stimulated and control B cells
b.interferon.response <- FindMarkers(immune.combined, ident.1 = "B_STIM", ident.2 = "B_CTRL", verbose = FALSE)
# Visualize expression of selected genes split by stimulation status
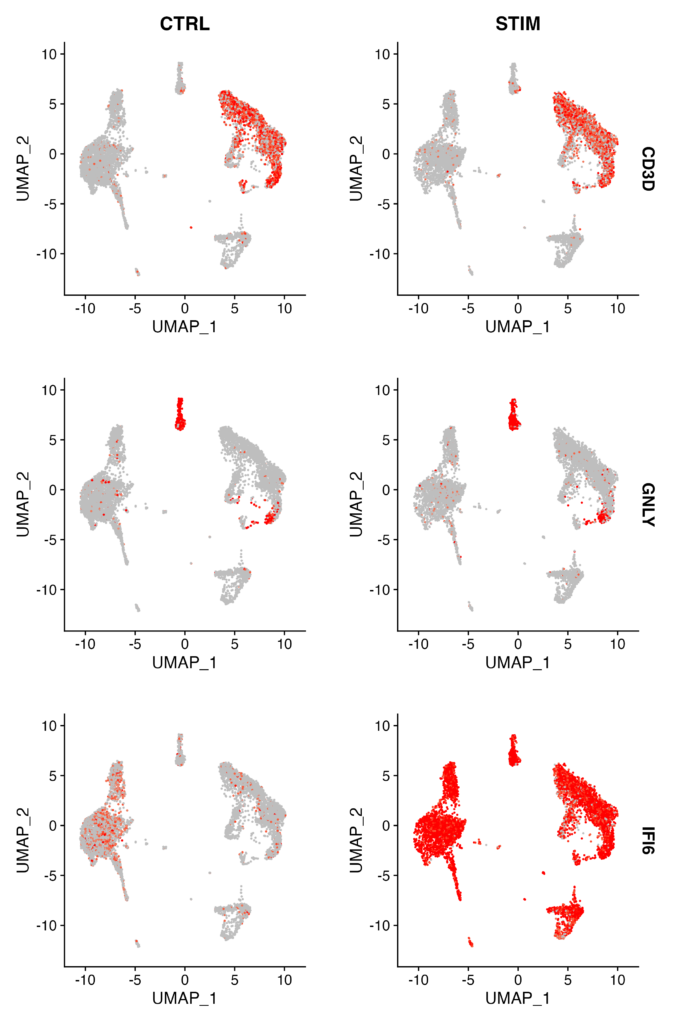
FeaturePlot(immune.combined, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3, cols = c("grey", "red"))
immune.combined$celltype.stimで、細胞のタイプと刺激の状態を組み合わせた新しい識別子を作成します。- 細胞の現在のタイプをバックアップし、新しい組み合わせた識別子で更新します。
FindMarkers関数を使用して、刺激されたB細胞とコントロールB細胞の間で差異的に発現している遺伝子を特定します。FeaturePlotを使用して、選択した遺伝子(CD3D、GNLY、IFI6)の発現を刺激の状態ごとに視覚化します。
以下のようなFeature plotが出れば成功です。CTRLとSTIMでどのクラスターで発現が変動しているか(以下だと、IFI6)一目瞭然ですね。
最後に各細胞種ごとの遺伝子発現をバイオリンプロットでも可視化してみましょう。
# Generate violin plots for specified genes, separated by stimulation status and grouped by cell type
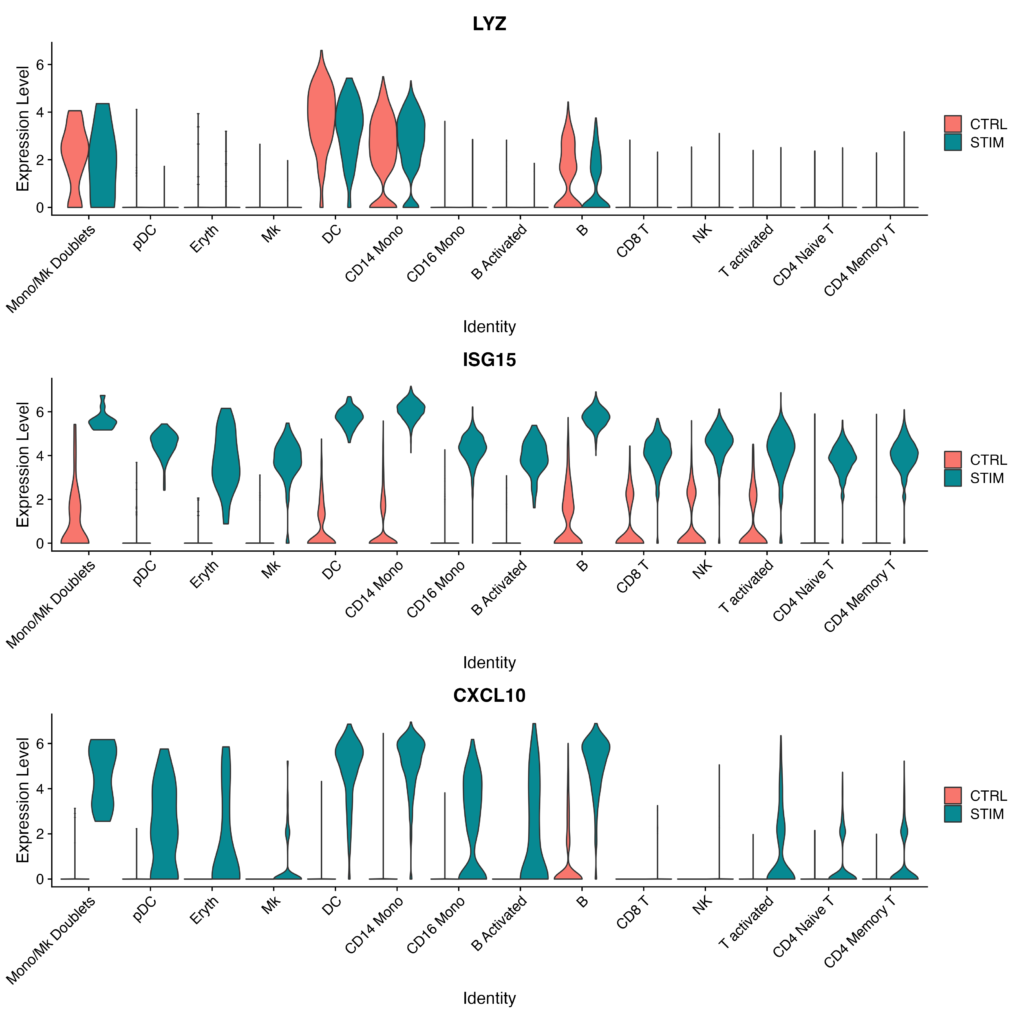
plots <- VlnPlot(immune.combined, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim", group.by = "celltype",
pt.size = 0, combine = FALSE)
# Display the generated plots in a single column layout
wrap_plots(plots = plots, ncol = 1)
- 指定された遺伝子(LYZ、ISG15、CXCL10)のバイオリンプロットを作成します。これらのプロットは、刺激の状態によって分割され、細胞タイプでグループ化されます。
- 作成されたバイオリンプロットを1列のレイアウトで表示します。
以下のように出力されれば成功です。ISG15やCXCL10は刺激後のサンプルにおいてExpression Levelが上昇しているのが各細胞種ごとにはっきりと可視化できてますね。
最後に
いかがだったでしょうか。scRNA-seqデータで複雑な細胞タイプに対する比較分析をコントロールと刺激サンプルで比較できるようになるだけで解析の幅がかなり広がったと思います。ぜひ挑戦してみてください。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

