
ローカル上でRNA-seq解析環境の整え方がわからない方はいませんか?また自分のPCが推奨環境のものではなく、環境依存のエラーに困っている方はいませんか?
そのような方に向けてブラウザ上でRNA-seq解析データの定量ができるRaNA-seqをご紹介いたします。
RaNA-seqを使える様になると自分のPC上でRNA-seq環境を整える必要がなく環境依存エラーの解決をしなくても良くなります。是非やってみましょう!
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
RaNA-seqとは?
RaNA-Seqは、RNA-Seqデータを迅速に解析するためのオープンなバイオインフォマティクスツールです。FASTQファイルの定量、品質管理メトリクスの計算、差次的発現解析の実行、機能解析による結果の解釈を可能にする完全な解析を数分で実行します。手持ちのFastqファイルをRNA-seq解析の複雑な環境構築なしでブラウザ上で実行できることで定評があります。またヒトやマウスに限らず、多種多様なレファレンスで定量できるのもポイントです。
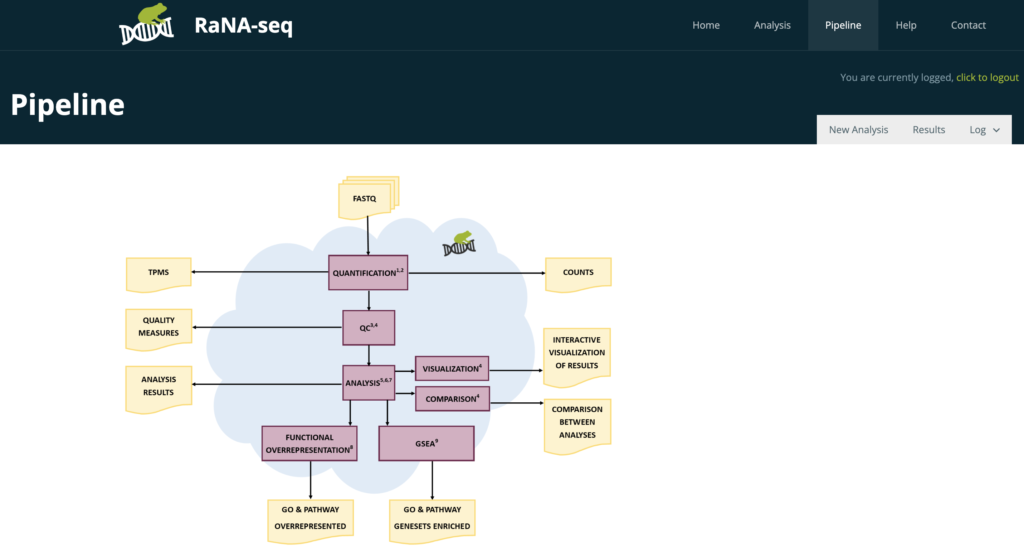
公式のホームページにてRaNA-Seqの主な特徴がまとめられています。
- FASTQファイルから結果の機能解析までの完全なRNA-Seq解析
- ソフトウェアのインストールが不要なスタンドアローン・ウェブサイト
- ENAリポジトリとの接続
- 非経験者向けの入力パラメータの自動設定
- 複数のグラフとメトリクスによる入力サンプルの品質管理
- 入力パラメータと複数の差分発現メソッドによる解析のカスタマイズ
- 結果の機能的過剰代表分析
- 遺伝子セット濃縮解析(GSEA)結果
- 分析に関する完全な情報を記載した報告書を作成し、出版物に掲載できるようにする
- インタラクティブなグラフィックによる結果のプレゼンテーション
- 一般に認められた再現可能なプロトコル
今回は、RaNA-seqを使ってリードカウントを取得する方法にフォーカスして解析していきます。その後の下流解析はまた別の記事にて紹介します。
fastq.gzファイルをダウンロードする
fastqファイルをダウンロードするにはfasterq-dumpを使用します。
fasterq-dumpを使うにはSRA-toolkitが必要なのでまだの方はこちらを参考にダウンロード&設定をお願いします。
fasterq-dumpでダウンロードしていきます。以下のサンプルコマンドのSRR番号を変更してターミナルを開き入力します。SRR番号は何個連ねて書いても大丈夫ですが、ネットワークエラーでよく落ちることがあるのでこまめにダウンロード状況を確認するようにしてください。
for accession in **SRR16922242 SRR16922243 SRR16922244**; do
fasterq-dump --split-files --progress $accession
gzip ${accession}*.fastq
done
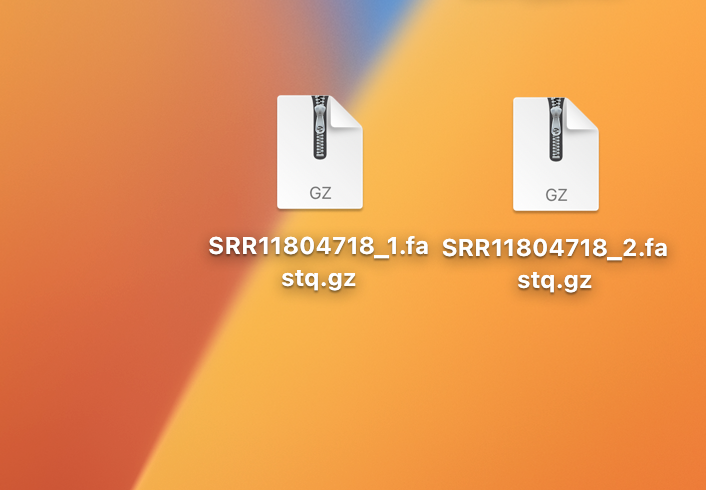
ダウンロードが完了すると以下のように、SRR〇〇_1.fastq.gz, SRR〇〇_2.fastq.gz という2つのファイルができます。
RaNA-seqの使い方
それでは実際にRaNA-seqを使ってみましょう。最初はRaNA-seqのアカウントを作成する必要がありますので、こちらからアクセスして作成してください。フォームに入力すればすぐアカウント作成ができます。
アクセスしたら「Analysis」タブで移動します。
1) Sample typeではレファレンスとなるゲノムを選びます。自分が対象としている生物や植物を選んでくださ
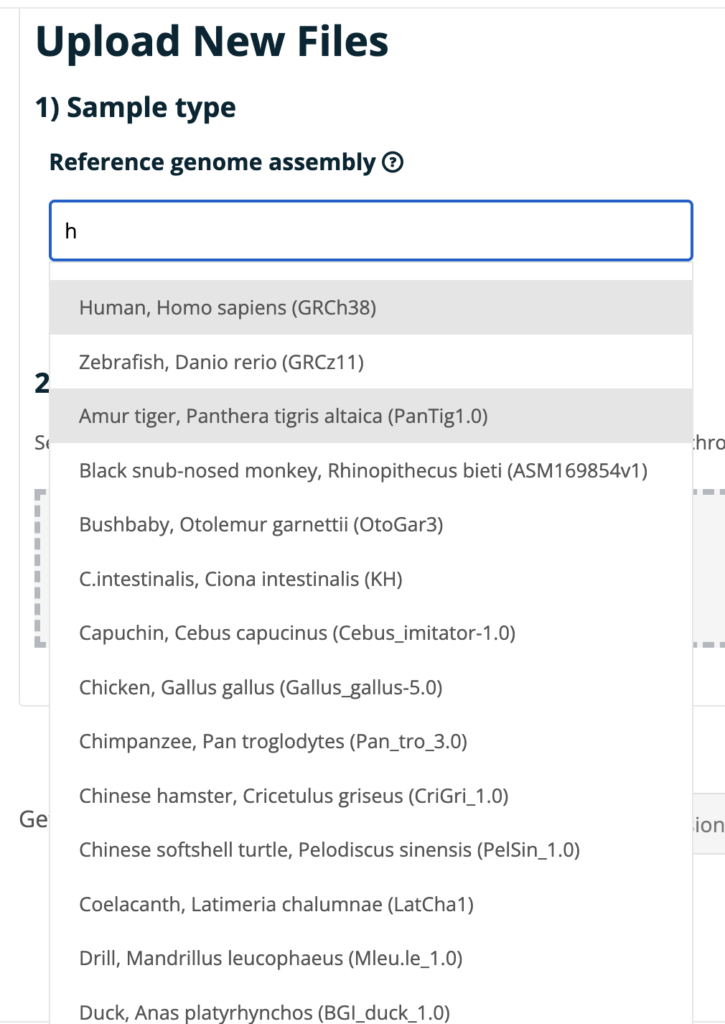
2)Select your FASTQ Filesでアップロードしていきます。
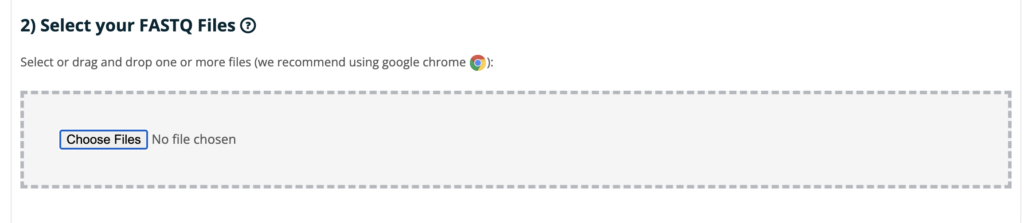
Paired-endの方は_1と_2の両方をアップロードすることを忘れないでください。
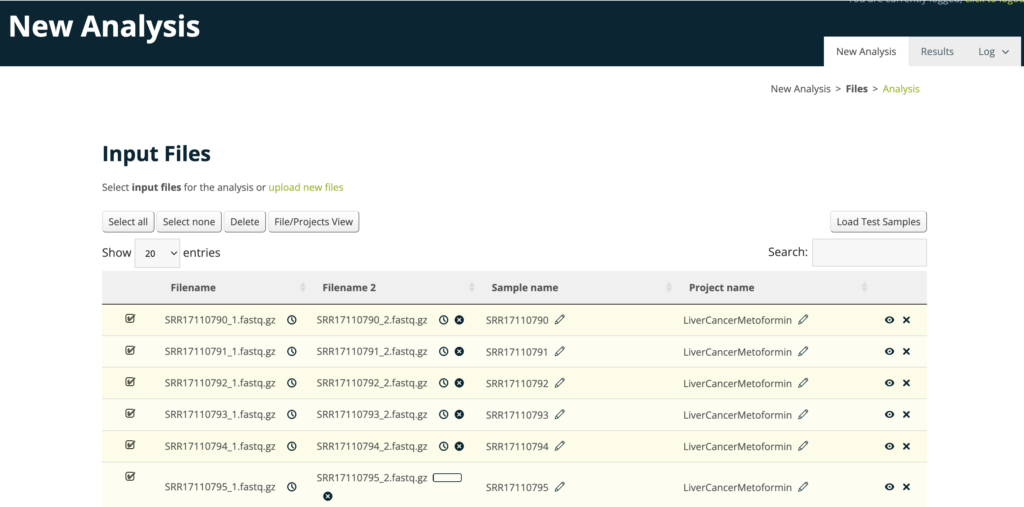
アップロードすると以下のような画面になります。
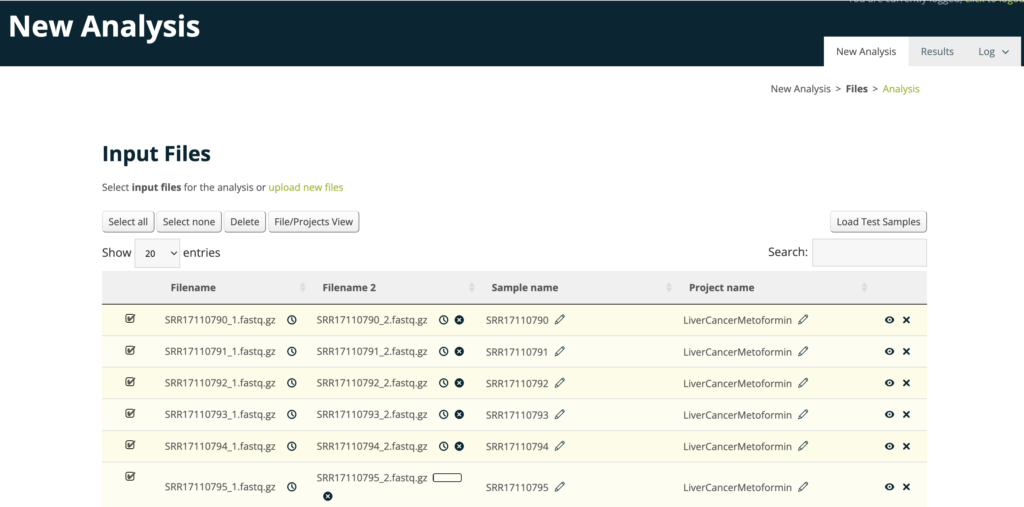
サンプルが用意できない方は、Load Test Samplesでサンプルファイルをロードできるので、お試しにやってみるといいと思います。
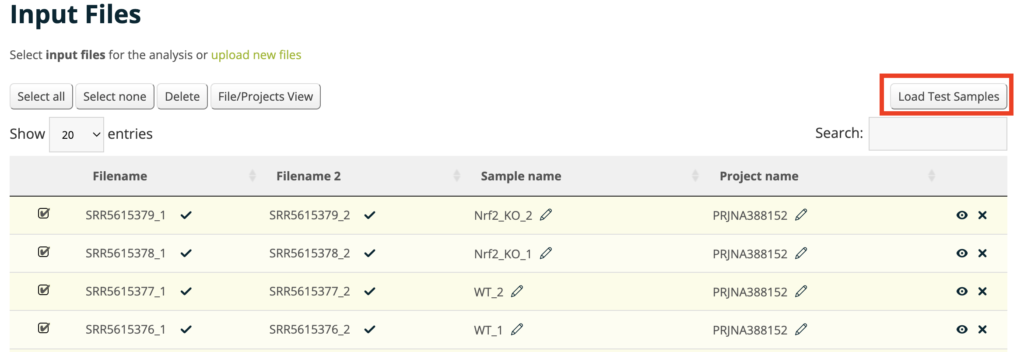
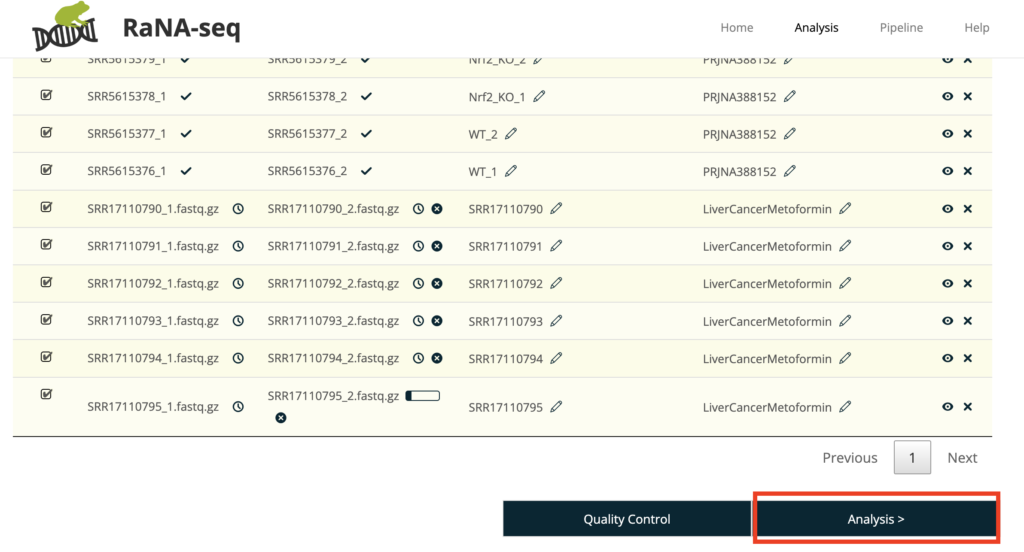
それでは右下から「Analysis」に進みます。
この画面は、新規解析セットアップ画面です。コントロール群とサンプル群の間でRNAの発現差を分析するためにデータセットをセットアップしていきます。③のようにまずはサンプルをコントロール群とサンプル群にドラック&ドロップで振り分けてください。
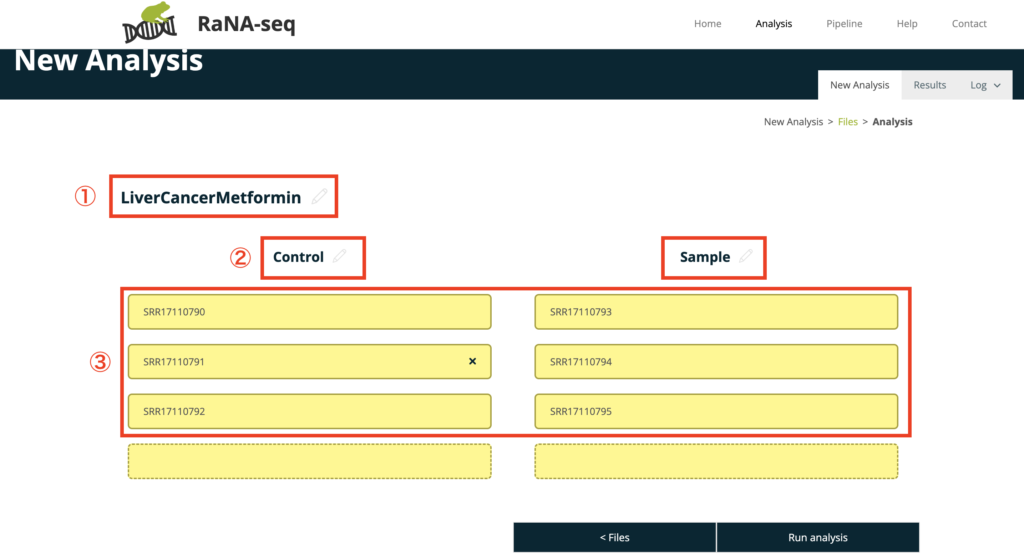
- 解析名の入力:この部分では、解析に名前を付けています。ここでの「LiverCancerMetformin」という名前をつけています。
- コントロール群の選択:「Control」と記載された部分には、シーケンスリードアーカイブ(SRA)の識別子が入力されています(例: SRR17110790)。これらは実験のコントロール群として使用されるデータセットを指しています。
- サンプル群の選択:同様に、「Sample」と記載された部分には、実験群または処理群のデータセットのSRA識別子が入力されています(例: SRR17110793, SRR17110794, SRR17110795)。
全てのデータセットが正しく入力された後、「Run analysis」ボタンをクリックして計算処理を開始し、RNAの量を定量化し、品質管理と差異発現解析などを行います。その結果から、コントロール群と処理群の遺伝子発現プロファイルの違いを明らかにすることができます。

ここまでできたら、解析をセット完了です。
RaNA-seqの結果の参照
それでは実際に解析結果を確認してみましょう。
右上の「Results」をクリックします。
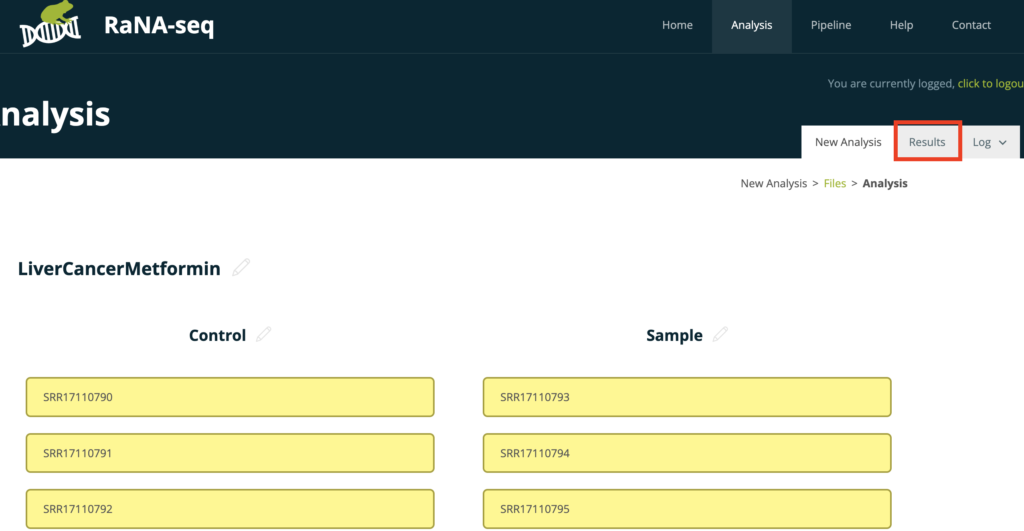
解析が終わってなければ、各ステップがwaitingにっていますので待ちましょう。
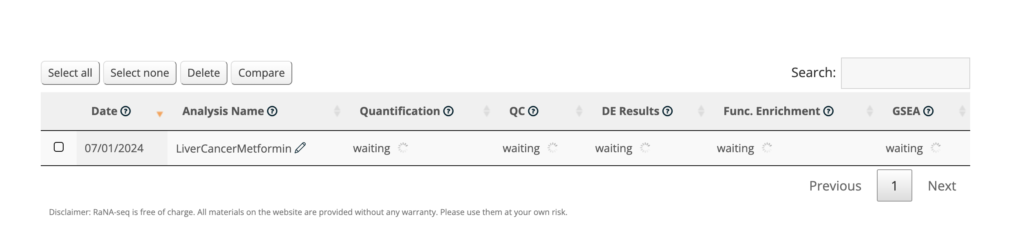
下記のように解析が終わったステップはstatusが「done」になります。doneの横のマークをクリックすると解析結果を得る事ができます。TPMデータなどを得るにはQuantificationの項目をクリックしてみましょう。
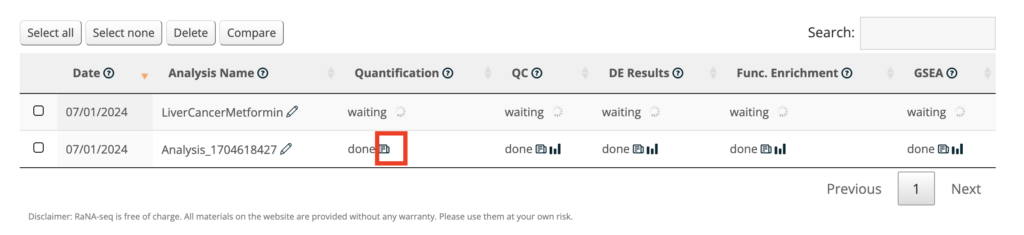
下流の解析にはTPMsのTSVなどを用いればよいです。
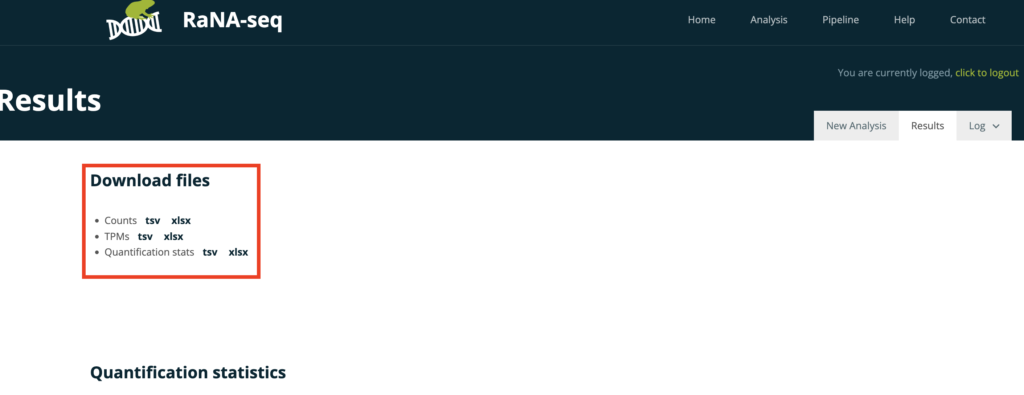
これで解析が完了しました。
最後に
いかがだったでしょうか。RaNA-seqを使えば、環境構築なしでTPMデータまで手に入るのは大変便利です。ぜひ習得してみてください。

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!

