
本記事はAI創薬の一つである機械学習を用いたin silico screeningについて書かれた記事です。第5章まであり、すべての内容が理解できると、目的の標的にあった薬物候補化合物を発見することができます。こちらの記事は第3章で、第1章と第2章で取得したデータの整形を行います!データサイエンティストっぽい泥臭い作業ですが、コピペでできるので、ぜひ、トライしてみてください!
macOS Ventura(13.2.1), Google colaboratory
今回は直ぐにpythonが実行できるようにGoogle colabを利用していきます。

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1500 今なら50%OFF!!
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1,500 今なら50%OFF!!
分子ドッキングやMDシミュレーションなど、
自宅でできるin silico創薬の解析方法を解説したものになります!
AI創薬とは?
AI創薬は、人工知能(AI)技術を利用して新しい薬物を発見、開発するプロセスです。AIは大量のデータを高速に処理し、薬物の候補を予測したり、薬物相互作用を評価したりします。また、AIは薬物の効果や安全性をシミュレートすることも可能で、臨床試験の前の段階でリスクを評価することができます。これにより、薬物開発のコストと時間を大幅に削減することが期待されています。AI創薬は、薬物開発の新しいパラダイムとして注目を集め、製薬企業や研究機関で積極的に研究、導入が進められています。また、バイオインフォマティクス、ケモインフォマティクス、機械学習、ディープラーニングなどの技術が組み合わされ、薬物開発のプロセスを革新しています。さらに、AI創薬は個人化医療の推進にも寄与し、患者にとって最適な治療法を提供する可能性を秘めています。
今回はAI創薬の中でも、in silico screeeningに焦点を当てて解説してきます。
以下の論文、記事をフォローしていきます。
NCBI – WWW Error Blocked Diagnostic
論文と公共データベースを使って無料で始めるAI創薬 – Qiita
この論文では新型コロナウイルスの標的タンパク質である3C-like プロテアーゼ(メインプロテアーゼ:Mpro)に対して、公共データベースにある化合物でスクリーニングを行い、最終的に41個のの化合物を薬物候補化合物としています。
AI創薬によるIn silico screeningの流れ
以下のような流れで行っていきます。全5章となります。
- 公共データベース(ChEMBL)からの機械学習の学習データを収集
- スクレイピングによる公共データベース(PDB)からの機械学習の学習データを収集
- 機械学習の学習データの整形(本記事)
- 整形データを用いた予測モデルの作成
- 予測モデルによる候補化合物のin silico screening
CheEMBLとPDBデータのマージ
ここではまず第1章と第2章で生成したデータをマージしていきます。
まずは下準備として、ライブラリや簡単なリストの生成、パスの指定を行います。
以下を実行してください。
!pip install rdkit-pypi
import csv
from collections import defaultdict
import pandas as pd
from rdkit import Chem
def smiles2smiles(smiles):
mol = Chem.MolFromSmiles(smiles)
return Chem.MolToSmiles(mol)
このコードは、化学構造を表すSMILES(Simplified Molecular Input Line Entry System)形式の文字列を取得し、それをRDKitライブラリを用いて処理し、再びSMILES形式の文字列として返すPython関数 smiles2smiles を定義しています。それぞれのステップについて説明します。
!pip install rdkit-pypi: この行は、Pythonパッケージ管理システムであるpipを用いて、**rdkit-pypiパッケージをインストールしています。rdkit**は、化学情報学と機械学習に広く使われるオープンソースのツールキットです。この行は、特にJupyterノートブックやGoogle Colabのような環境で実行されます。import csv: ここでは、Pythonの標準ライブラリであるcsvモジュールをインポートしています。このモジュールは、CSV(コンマ区切り値)ファイルの読み書きに使われます。from collections import defaultdict: ここでは、Pythonの標準ライブラリであるcollectionsモジュールからdefaultdictをインポートしています。defaultdictは、辞書(dictionary)と似ていますが、キーが存在しない場合にデフォルト値を自動的に生成する機能があります。import pandas as pd: ここでは、データ分析に広く使われるPandasライブラリをインポートし、**pd**という略称で使用するための宣言をしています。from rdkit import Chem: RDKitのChemモジュールをインポートしています。このモジュールは、化学構造を操作するための関数やクラスを提供します。def smiles2smiles(smiles):: **smiles2smiles**という関数を定義しています。この関数は、SMILES形式の文字列を引数として取ります。mol = Chem.MolFromSmiles(smiles): 引数で与えられたSMILES文字列から、RDKitの分子(Molオブジェクト)を生成します。return Chem.MolToSmiles(mol): 生成されたMolオブジェクトをSMILES形式の文字列に変換して返します。このステップは、元のSMILES文字列を構造的に正確な形式に変換するために役立ちます。
**smiles2smiles(smiles)**関数の目的は、与えられたSMILES文字列をRDKitを使って構造的に正規化し、その結果を再びSMILES形式で出力することです。これにより、元のSMILES文字列の構造的なエラーや不規則性を修正することができます。
続いて、各ファイルをいれ、以下を実行してください。
最初はprocessed_compounds_of_details.csv と3C-like_and_main_proteases.csv の二つがアップロードしている状態です。
各ファイルのパスの定義と空のリストの定義を行っています。
input_chembl_path = "/content/processed_compounds_of_details.csv"
input_pdb_path = "/content/3C-like_and_main_proteases.csv"
output_file_path = "/content/output.csv"
merged_file_path = "/content/merged_data.csv"
ids = list()
org_smiles = list()
canonical_smiles = list()
outcomes = list()
続いて、以下を実行してください。
# ChEMBLデータの読み込み(chembl_id, smiles, 阻害値)
with open(input_chembl_path, "r") as f:
reader = csv.DictReader(f)
for row in reader:
ids.append(row["chembl_id"])
org_smiles.append(row["canonical_smiles"])
canonical_smiles.append(smiles2smiles(row["canonical_smiles"]))
outcomes.append(row["outcome"])
このコードのセクションは、CSVファイルからChEMBLデータを読み込み、特定の情報をリストに格納するために使われています。それぞれのステップについて詳しく見ていきます。
- CSVファイルの読み込み:
with open(input_chembl_path, "r") as f:この行は、**input_chembl_path変数に保存されているパスのCSVファイルを読み込みモードで開きます。with**文は、ファイルを開いた後に自動的に閉じるために使われます。
- CSVデータの読み込み:
reader = csv.DictReader(f)ここで、CSVファイルの内容を辞書形式で読み込むための**csv.DictReaderオブジェクトを作成しています。DictReader**は、各行をキーが列ヘッダー、値がその行のデータである辞書として読み込みます。
- データの抽出とリストへの追加:
for row in reader:このループは、CSVファイルの各行に対して反復処理を行います。ids.append(row["chembl_id"]): **chembl_id列のデータをids**リストに追加します。org_smiles.append(row["canonical_smiles"]): **canonical_smiles列のデータをorg_smiles**リストに追加します。canonical_smiles.append(smiles2smiles(row["canonical_smiles"])): **canonical_smiles列のデータをsmiles2smiles関数で処理して、その結果をcanonical_smiles**リストに追加します。この処理は、元のSMILES文字列を構造的に正規化するために行われます。outcomes.append(row["outcome"]): **outcome列のデータをoutcomes**リストに追加します。
このコードは、ChEMBLデータからの化合物のデータを読み込み、それを後の処理や分析のために整理することを目的としています。
続いて、以下のコードを実行してください。
# PDBデータを読み込み、阻害値で結果を生成
with open(input_pdb_path, "r") as f, open(output_file_path, "w", newline='') as outfile:
reader = csv.DictReader(f)
fieldnames = reader.fieldnames + ['outcome'] # 既存の列名に 'outcome' 列を追加
writer = csv.DictWriter(outfile, fieldnames=fieldnames)
writer.writeheader()
for row in reader:
# 'inhibitory_activity_value' の値をチェックし、条件に基づいて 'outcome' を設定
if int(row['inhibitory_activity_value']) >= 10000:
row['outcome'] = 0
else:
row['outcome'] = 1
writer.writerow(row)
# 結果を加えたPDBデータの読み込み(pdb_ligand_id, smiles, 阻害値)
with open(output_file_path, "r", encoding='utf-8-sig') as f:
reader = csv.DictReader(f)
print(reader.fieldnames) # 列名を印刷
for row in reader:
ids.append(row["ligand_id"])
org_smiles.append(row["smile"])
canonical_smiles.append(smiles2smiles(row["smile"]))
outcomes.append(row["outcome"])
このコードは、PDBデータにoutcome列がなかったので、それを追加することで、新しいCSVファイルを作成し、その後で加工されたデータをさらに読み込むというプロセスを実行しています。それぞれのステップを詳しく見てみましょう:
PDBデータの読み込みと加工
- ファイルの読み込みと書き出しの準備:
with open(input_pdb_path, "r") as f, open(output_file_path, "w", newline='') as outfile:この行は、入力となるPDBデータファイル(input_pdb_path)を読み込みモードで、出力ファイル(output_file_path)を書き込みモードで開いています。**newline=''**は、CSVの書き込みにおいて行間の余分な改行を防ぐために使用されます。
- CSVリーダーとライターの設定:
reader = csv.DictReader(f)でCSVファイルの内容を辞書形式で読み込んでいます。fieldnames = reader.fieldnames + ['outcome']で既存の列名に新しい列**'outcome'**を追加しています。writer = csv.DictWriter(outfile, fieldnames=fieldnames)でCSVファイルの書き込みの準備をしています。これにより、新しいデータの各行が辞書形式で書き込まれます。
- データの加工と新しいファイルへの書き込み:
- ループ内で、**
'inhibitory_activity_value'列の値を基にして'outcome'**列の値を設定しています。 - **
row['outcome'] = 0またはrow['outcome'] = 1とすることで、特定の条件(ここではinhibitory_activity_value**が10000以上かどうか)に基づいた結果を行に追加します。 - **
writer.writerow(row)**で加工された各行を新しいCSVファイルに書き込みます。
- ループ内で、**
加工されたPDBデータの読み込み
- 加工されたデータファイルの読み込み:
- 新しく作成されたCSVファイル(
output_file_path)を読み込みモードで開いています。 - ここでも**
csv.DictReader**を使用して、ファイルの内容を辞書形式で読み込みます。
- 新しく作成されたCSVファイル(
- データの抽出とリストへの追加:
- ループを使って、
ligand_id、smile、outcomeの各列からデータを取得し、それぞれids、org_smiles、canonical_smiles、**outcomes**リストに追加しています。 - **
smiles2smiles**関数を使用して、SMILES文字列を正規化しています。
- ループを使って、
このコードは、PDBデータから特定の情報を抽出し、加工して新しいフォーマットで保存し、その後で加工されたデータを更に分析や処理のために使用する流れを示しています
続いて、以下のコードを実行してください。
# pandasに変換
df = pd.DataFrame(data={"id": ids, "original_smiles": org_smiles, "canonical_smiles": canonical_smiles, "outcome": outcomes})
#重複チェック
# df["dup"] = df["canonical_smiles"].duplicated()
# インデックスを除外してCSVファイルに保存
df.to_csv("merged_file_path.csv", index=False)
このコードは、リストに格納されたデータを使ってPandas DataFrameを作成し、その後で重複をチェックし(現在はコメントアウトされている部分)、最終的にCSVファイルとして保存する処理を行っています。それぞれのステップを詳しく見ていきましょう。
Pandas DataFrameへの変換
df = pd.DataFrame(data={"id": ids, "original_smiles": org_smiles, "canonical_smiles": canonical_smiles, "outcome": outcomes}):- ここでは、**
pd.DataFrame**を使って新しいDataFrameを作成しています。 - **
data**引数に辞書を渡すことで、DataFrameの各列に対応するデータを指定しています。 ids,org_smiles,canonical_smiles,outcomesというリストがそれぞれ"id","original_smiles","canonical_smiles", **"outcome"**という列名に対応します。
- ここでは、**
重複チェック(コメントアウトされている)
# df["dup"] = df["canonical_smiles"].duplicated():- この行は、**
"canonical_smiles"**列内の重複した値をチェックするためのコードですが、コメントアウトされているため、実行されません。 - 実行されていれば、DataFrameに**
"dup"という新しい列を追加し、重複がある場合はTrue、ない場合はFalse**がその行に設定されることになります。
- この行は、**
CSVファイルへの保存
df.to_csv("merged_file_path.csv", index=False):- この行では、**
to_csv**メソッドを使ってDataFrameをCSVファイルとして保存しています。 - ファイル名は**
"merged_file_path.csv"ですが、ここでは変数merged_file_path**が使われていない点に注意してください。これは固定された文字列のファイル名です。 - **
index=False**は、保存するCSVファイルにDataFrameのインデックス(行番号)を含めないことを意味します。
- この行では、**
このコードにより、さまざまな情報を含むDataFrameがCSVファイルとして保存されます。保存されたデータは、後でのデータ分析や他の処理のために使用することができます。重複チェックの部分を実行する場合は、コメントアウトを解除してください。また、ファイル名に変数を使用する場合は**"merged_file_path.csv"の部分をmerged_file_path**に置き換えてください。
構造データからのフィンガープリントの作成
機械学習に応用するためには各構造をベクトルデータに変換する必要がある。この章ではその変換について記載します。
まずは必要なライブラリのインストールとインポートをします。
# 必要なライブラリをインストール
!pip install rdkit-pypi molvs
# ライブラリのインポート
from rdkit import Chem
from rdkit.Chem import AllChem
from molvs.normalize import Normalizer, Normalization
from molvs.tautomer import TautomerCanonicalizer
from molvs.fragment import LargestFragmentChooser
from molvs.charge import Uncharger
import pandas as pd
import numpy as np
ライブラリのインストール
!pip install rdkit-pypi molvs:- この行はPythonのパッケージインストーラーであるpipを使って、**
rdkit-pypiとmolvs**という二つのパッケージをインストールしています。 - **
rdkit-pypi**は化学情報学の分野で広く使用されるRDKitライブラリのPythonパッケージです。分子の操作、構造の分析、化学的特性の計算などに使用されます。 - **
molvs**は、化合物の正規化、標準化、荷電状態の調整などを行うためのツールキットです。RDKitと組み合わせて使用されることが多いです。
- この行はPythonのパッケージインストーラーであるpipを使って、**
ライブラリのインポート
from rdkit import Chemとfrom rdkit.Chem import AllChem:- RDKitライブラリから、**
ChemモジュールとAllChem**サブモジュールをインポートしています。これらは分子の生成、操作、および化学的特性の計算に使われます。
- RDKitライブラリから、**
from molvs.normalize import Normalizer, Normalization:- **
molvsライブラリから、化合物の正規化に使用されるNormalizerクラスとNormalization**クラスをインポートしています。
- **
from molvs.tautomer import TautomerCanonicalizer:- 分子のタウトマー(化学構造の異なる形態)の正準形を取得するための**
TautomerCanonicalizer**クラスをインポートしています。
- 分子のタウトマー(化学構造の異なる形態)の正準形を取得するための**
from molvs.fragment import LargestFragmentChooser:- 複数のフラグメント(分子の断片)が存在する場合に最大のフラグメントを選択するための**
LargestFragmentChooser**クラスをインポートしています。
- 複数のフラグメント(分子の断片)が存在する場合に最大のフラグメントを選択するための**
from molvs.charge import Uncharger:- 分子の荷電状態を調整するための**
Uncharger**クラスをインポートしています。
- 分子の荷電状態を調整するための**
import pandas as pdとimport numpy as np:- データ分析に広く使用されるPandasライブラリとNumPyライブラリをインポートしています。Pandasはデータフレームの操作、NumPyは数値計算や配列の操作に使用されます。
これらのライブラリは、化学データの処理、分析、および可視化に不可欠であり、化学情報学、薬学、生物学の分野で広く利用されています。
続いて、以下を実行してください。
def normalize(smiles):
# Generate Mol
mol = Chem.MolFromSmiles(smiles)
# Uncharge
uncharger = Uncharger()
mol = uncharger.uncharge(mol)
# LargestFragmentChooser
flagmentChooser = LargestFragmentChooser()
mol = flagmentChooser(mol)
# Sanitaize
Chem.SanitizeMol(mol)
# Normalize
normalizer = Normalizer()
mol = normalizer.normalize(mol)
tautomerCanonicalizer = TautomerCanonicalizer()
mol = tautomerCanonicalizer.canonicalize(mol)
return Chem.MolToSmiles(mol)
この**normalize**関数は、化学構造を表すSMILES文字列を取得し、RDKitとMolVSライブラリを使用して一連の処理を行い、正規化されたSMILES文字列を返します。それぞれのステップを詳しく見ていきましょう:
- 分子の生成 (
Chem.MolFromSmiles(smiles)):- 引数で与えられたSMILES文字列からRDKitの分子オブジェクト(Molオブジェクト)を生成します。
- 荷電状態の除去 (
Uncharger()):- **
Uncharger**クラスのインスタンスを作成し、分子から不必要な荷電を取り除きます。これは、分子をより一般的な状態にするために重要です。
- **
- 最大フラグメントの選択 (
LargestFragmentChooser()):- 複数のフラグメントが存在する場合に最大のフラグメントを選択します。これにより、不要な小さなフラグメントが除去されます。
- 分子の清浄化 (
Chem.SanitizeMol(mol)):- 分子を清浄化(sanitize)し、化学的に意味のある状態にします。この処理により、芳香族性の判定や原子価のチェックなどが行われます。
- 分子の正規化 (
Normalizer()):- **
Normalizer**クラスを使って分子を正規化します。これにより、分子内の特定の変換が適用され、より標準化された形式になります。
- **
- タウトマーの正準化 (
TautomerCanonicalizer()):- **
TautomerCanonicalizer**クラスを使って分子のタウトマーを正準化します。異なるタウトマー形態の中から最も代表的または最も安定した形態を選択します。
- **
- 正規化されたSMILESの生成と返却 (
Chem.MolToSmiles(mol)):- 最終的に、処理された分子を再びSMILES形式に変換し、この正規化されたSMILES文字列を返します。
この関数は、化学データベースの構築、化学構造の分析、または機械学習モデルのためのデータ前処理において非常に有用です。正規化プロセスにより、データの一貫性が保たれ、分析の信頼性が向上します。
最後に以下のコードを実行してください。
import csv
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem
import numpy as np
# 学習データの読み込み
input_path = "/content/merged_file_path.csv"
fingerprint_path = "/content/fingerprint.csv"
datas = [] # すべてのデータを保持するリスト
with open(input_path, "r") as f:
reader = csv.DictReader(f)
for row in reader:
org_smile = row["canonical_smiles"]
new_smile = normalize(org_smile)
mol = Chem.MolFromSmiles(new_smile)
fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=3, nBits=2048, useFeatures=False, useChirality=False)
fp = pd.Series(np.asarray(fp)).values
data = []
data.append(row["id"])
data.append(int(row["outcome"]))
data.extend(fp)
datas.append(data)
# ループの外でDataFrameの作成
columns = ["id", "outcome"] + [f"Bit_{i+1}" for i in range(2048)]
df = pd.DataFrame(datas, columns=columns)
df.set_index("id", inplace=True, drop=True)
# 一度だけファイルに保存
df.to_csv(fingerprint_path)
このコードは、CSVファイルから学習データを読み込み、RDKitを使用して化合物のSMILES表現を正規化し、Morganフィンガープリントを生成し、最後にPandas DataFrameにこれらのデータをまとめてCSVファイルとして保存する一連の処理を行っています。それぞれのステップについて説明します。
CSVファイルの読み込みとデータ処理
- ファイルの読み込み:
- **
input_pathからCSVファイルを読み込み、datas**リストを初期化しています。このリストは処理されたデータを格納するために使用されます。
- **
- CSVデータの読み込みと処理:
- CSVファイルから各行を読み込み、**
normalize関数を使ってcanonical_smiles**列のデータを正規化しています。 - RDKitの**
Chem.MolFromSmilesとAllChem.GetMorganFingerprintAsBitVect**関数を使用して、正規化されたSMILESからMorganフィンガープリントを計算しています。
- CSVファイルから各行を読み込み、**
- データの集約:
id、**outcomeの値と計算されたフィンガープリントをdata**リストに追加しています。- その後、**
datasリストにこのdata**リストを追加しています。
DataFrameの作成とCSVファイルへの保存
- DataFrameの作成:
- **
datas**リストを使用してPandas DataFrameを作成しています。 - DataFrameの列名は、
id、outcome、そして2048個のフィンガープリントのビットごとの列名(Bit_1,Bit_2, …,Bit_2048)です。
- **
- DataFrameの保存:
- **
df.to_csv(fingerprint_path)**を使って、DataFrameをCSVファイルに保存しています。
- **
このコードは、化学データの前処理と分析のための基本的なフローを示しています。このプロセスを通じて、化合物の特徴を表すフィンガープリントが生成され、それを含むデータがさらに分析や機械学習のために利用可能な形式に変換されます。DataFrameの使用は、データの操作と保存を簡単にするための効果的な方法です。
以下のようなファイルが生成すれば完了です!
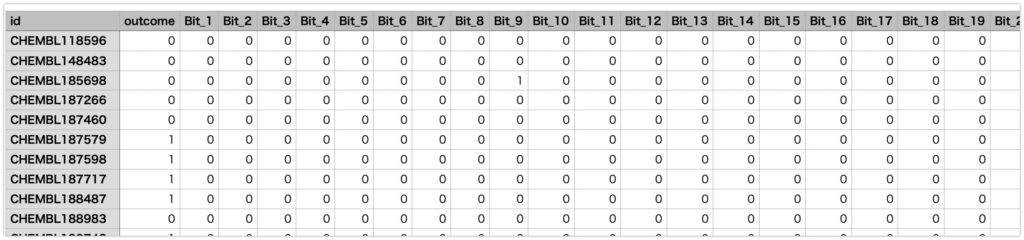
最後に
いかがでしたでしょうか?データの変換なので、今回は非常に泥臭い作業でしたが、構造式をsmileに直したり、Morganフィンガープリントを生成したりと非常にケモインフォマティクスをやっている感じを体験できたと思います!これからは第4章ではこれらのデータを使って機械学習モデルを作成していきます。
参考文献
RDkitについて詳しく知りたい方
Morganフィンガープリントについて詳しく知りたい方
【RDKit】Morganフィンガープリントの生成ルールを解釈してみた – Qiita
謝辞
この記事は以下の論文のフォローとなります。機械学習とAI創薬についてわかりやすく書かれています。このような論文を書いて下った筆者らに感謝を申し上げます。
NCBI – WWW Error Blocked Diagnostic
この記事を書くにあたり、**@kimisyo(晶 公畑)**さんの記事が大変参考になりました。ありがとうございます。

