
RNA-seq解析の手法の一つにメタ解析と呼ばれる手法があることを聞いたことがありますでしょうか。メタ解析はNGSを持っている研究室だけができる研究手法でしょ?と思っている人もいるかも知れませんが、最近は公共データを大量に解析することで、データを再活用することも注目されています。
この記事では、RNA-seq解析自動化パイプラインであるikraを用いて、論文で使われているようなメタ解析の手法をご紹介したいと思います。解析の時間はかかりますが、難しくありません。メタ解析にはいろいろなやり方がありますが、今回はikraと相性のよいメタ解析のやり方を紹介します。
メタ解析を学べば、それだけで論文が書けてしまうほど強力な手法になります。ぜひ身につけていきましょう!
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
メタ解析とは
メタ解析は、大量のサンプルの中から共通して変動がある物質に注目する手法です。
トランスクリプトームの文脈では、様々な研究室で得たサンプル(バックグラウンドが異なると言います)を大量に用意して、RNA-seqによって遺伝子発現量を定量、共通した変動遺伝子(DEG)を抽出して解析する手法です。
十分にサンプル量があり、そのどれも共通したDEGがある場合は、統計的に意味のある変動である可能性が高くなります。
ikraとは
ikraはRNA-seqの生データから、RNA-seq解析を完全自動化したツールになります。
RNA-seq解析を行うには複数のツールを組み合わせて段階的に解析していく必要がありますが、ikraではすべてのツールをパイプラインとしてつなぐことで、実行コマンド一つで自動的に各ツールが実行されていく仕組みになっています。
詳しくはこちらの【バイオインフォマティクス】公共データを用いて最速でRNA−seq解析を行う方法 【ikra】でikraの使い方を紹介しておりますので、よろしければご参照ください。
メタ解析のやり方
メタ解析に必要なデータを集める
まずはキーワードを決めてSRAデータを集めてくる作業を行います。
メタ解析では特定のキーワードを決めることが大事です。
自分の研究に関連があるキーワードを思い浮かべましょう。
例えば、酸化ストレスや低酸素応答に関連するキーワードは以下が挙げられます。
oxidative stress, rotenone, paraquat, hydrogen peroxide (H2O2),
UV, lipopolysaccharide, arsenite, deoxynivalenol,SRAデータを集めるサイトにはAOEを利用するのがおすすめです。NCBIやDBCLSでも問題はありません。自分の扱いやすいサイトをお選びください。
AOEの使い方は以下のページで紹介しておりますので、ご参照ください。APIで簡単に引っ張ってくる方法も紹介しております。
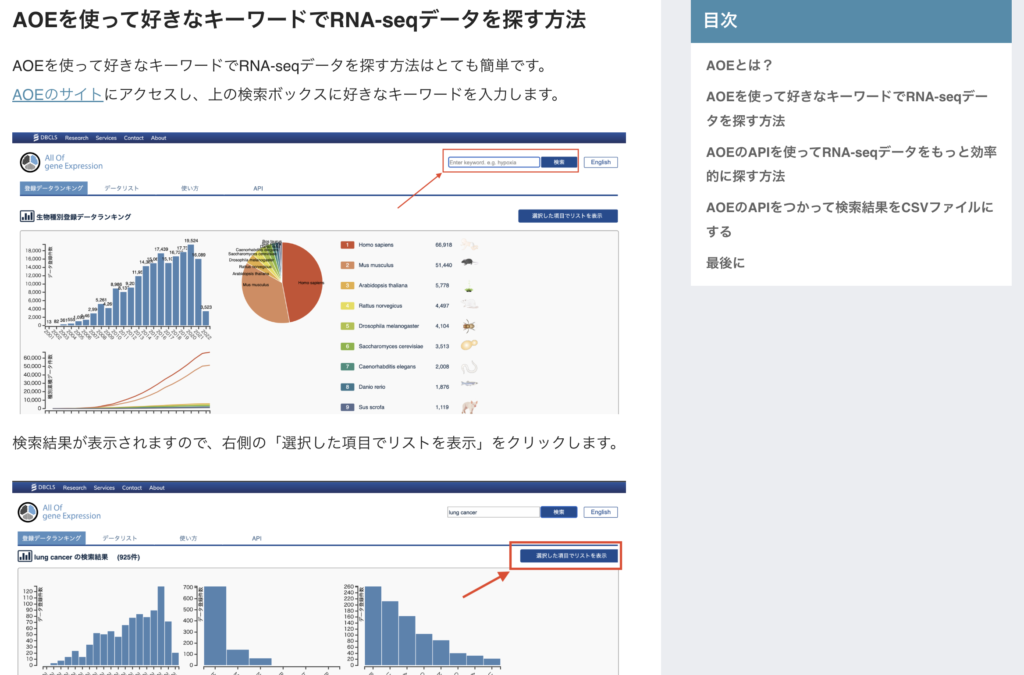
メタ解析に必要なデータ数ですが、多ければ多いほどよいですが、100は最低でもほしいところです。
マイナーなキーワードは他のキーワードを含めるか、もう少し研究を大きく捉えて(「ある特定の抗がん剤」ではなく、「癌」についてなど)キーワードを設定するのがコツです。
検索結果をみてどれだけサンプル数が用意できそうか、また特定の研究室のデータだけでなく、幅広いデータ種類を集められると良いと思います。
ikraを用いてTPMデータを作成する
キーワードが決まったら、キーワードをもとに出てきたSRAデータを用いてikraでどんどん解析していきましょう。
ikraを使うと、output.tsvというscaledTPMが記載されたファイルが得られます。output.tsvに対してまずは以下の操作をします。
- output.tsvの数値すべてに1を足す
- コントロール群を平均化する
- 刺激を与えたsample群を、2.の平均化されたコントロール群で割る(ratio)
式にすると以下の通りになります。
(sample +1) / ( control1…N +1)/N 1の操作をする理由は、発現量が0の場合は、 0で割ってしまう場合があるため、エラーにならないように1を足します。
3.の操作を行うことで、刺激前と刺激後の発現量のratioデータを出すことができるので、遺伝子間やサンプル間の発現量比較が可能になります。
ここまでの操作をすると以下のようなテーブルが出来上がっていくと思います。今一度ご確認ください。
| SRR11111111 | SRR11111112 | SRR11111113 | … | |
| A1BG | 1.204012121 | 1.305946294 | 1.058155716 | |
| A1CF | 0.8814023041 | 1.010914863 | 1.174708571 | |
| A2M | 1.868867371 | 1 | 0.9556968534 | |
| A2ML1 | 0.6709431377 | 1.126605147 | 1 | |
| A3GALT2 | 1 | 1 | 1 | |
| A4GALT | 0.5010093702 | 1.364249746 | 1.664916037 | |
| A4GNT | 1.881158763 | 1 | 1 | |
| AAAS | 0.7203903135 | 0.8684943431 | 1.474680837 | |
| AACS | 1.06245034 | 1.256530797 | 1.544221892 | |
| (以下略) |
ratioデータでは以下のような意味を持ちます。
- ratio > 1 のとき 発現上昇遺伝子
- ratio < 1 のとき 発現減少遺伝子
- ratio = 1 のとき 発現変動なし
これら遺伝子変動が有意的な変動かどうかは個々に用いられたsampleを用いて有意差検定をしなければもっともらしいことは言えませんが、メタ解析ではratioがいくつのときに発現変動とみなすかを検討するのも重要になってきます(ratio > 2のときに発現上昇遺伝子とする)。
次のscoreを決めるの項目で紹介するで、発現変動とみなす基準の一つを紹介します。
ratioデータをもとにscoreを決める
ratioデータだけでは解釈が難しいので、分かりやすい形にしていきましょう。
ここでは発現上昇遺伝子(ratio > 5 , 10)と発現減少遺伝子(ratio < 5 , 10)をそれぞれ、up5とup10、 dn5, dn10としてscoreはサンプル分カウントしていきます。
以下のような表を作っていけばよいでしょう。
| up5 | up10 | dn5 | dn10 | サンプル数 | score 5以上 | score 10以上 | |
| A3GALT2 | 1 | 0 | 0 | 0 | 34 | 1 | 0 |
| A4GALT | 1 | 0 | 0 | 0 | 34 | 1 | 0 |
| A4GNT | 0 | 0 | 0 | 0 | 34 | 0 | 0 |
| ABO | 5 | 3 | 7 | 0 | 34 | -2 | 3 |
| AGA | 0 | 0 | 0 | 0 | 34 | 0 | 0 |
| ALG1 | 0 | 0 | 0 | 0 | 34 | 0 | 0 |
| ALG10 | 0 | 0 | 1 | 1 | 34 | -1 | -1 |
| (以下略) |
scoreの列ですが、発現上昇サンプル数から発現減少サンプルを引いて算出します。
式にすると以下のようなイメージです。
score5以上 = up5のサンプル数 - dn5のサンプル数
score10以上 = up10のサンプル数 - dn10のサンプル数scoreの列を出したら、それを降順にすると遺伝子上昇が大きい順に、昇順にすると発現抑制が大きい順にサンプルが出てきます。
ここまで来たら後は、発現上昇が見られた上位5%など各自で閾値を設定して、発現変動が見られた遺伝子について考察すればメタ解析として成立します!
終わりに
いかがだったでしょうか。メタ解析はずっと難しいと思っていたけど、思ったよりも難しい解析をしていないという印象を抱いた方もいらっしゃるのではないでしょうか。今回は参考にした論文を記載しておきますので、実際の論文での事例を確認したい場合はそちらをご参照ください。
ikraを回す時間こそかかりますが、ウェット実験に取り組んでいる間に自分のPCで解析を回しておけば、効率よく研究を進められると思います。是非チャレンジしてみてください!
参考文献
- Ono, Y., & Bono, H. (2021). Multi-omic meta-analysis of transcriptomes and the bibliome uncovers novel hypoxia-inducible genes. Biomedicines, 9(5). https://doi.org/10.3390/biomedicines9050582
- Canton, M., Forestan, C., Bonghi, C., & Varotto, S. (2021). Meta-analysis of RNA-Seq studies reveals genes with dominant functions during flower bud endo- to eco-dormancy transition in Prunus species. Scientific Reports 2021 11:1, 11(1), 1–15. https://doi.org/10.1038/s41598-021-92600-6
- Suzuki, T., Ono, Y., & Bono, H. (2021). Comparison of Oxidative and Hypoxic Stress Responsive Genes from Meta-Analysis of Public Transcriptomes. Biomedicines, 9(12), 1830. https://doi.org/10.3390/biomedicines9121830
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

