
RNA−seq解析にエンリッチメント解析を用いる方は多いのではないでしょうか。DAVIDやMetascapeなどのブラウザツールを駆使すれば、エンリッチメント解析をするのは容易だと思います。
本記事ではR言語のパッケージであるclusterprofilerをもちいてエンリッチメント解析をする方法について解説します。
R言語を用いたエンリッチメント解析をすると、より柔軟なカスタマイズが可能となります。しかもそんなに難しく無いので、ぜひ挑戦してみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
エンリッチメント解析について
エンリッチメント解析とは、生物学的データを分析するための方法の一つです。この解析は、特定のバイオマーカーや遺伝子セットが指定された生物学的なパスウェイや生物学的なプロセスにどの程度高確率で関連しているかを求めることを目的としています。
この解析には、対象となる遺伝子セットが対象となるパスウェイや生物学的プロセスに対する期待値と比較して、統計的に有意に高い割合で含まれているかどうかを評価することが含まれます。このような解析により、生物学的なデータをより深く理解するための新しい仮説の発見や、既存の知識を検証するための手がかりを得ることができます。
R言語を用いたエンリッチメント解析は、様々ありますが、今回は”clusterProfiler”を紹介します。clusterprofilerを用いたエンリッチメント解析のメリットの一つに豊富な可視化手段があります。 clusterprofilerには多くのグラフ作成用の関数が用意されており、エンリッチメント解析の結果を視覚的に表現することができます。
遺伝子名を表し方
cluterprofilerを使うにあたって、遺伝子名の表し方にはどのようなものがあるかを確認しておきましょう。今回は皆さんにも馴染みがよくある形としてSYMBOL型があると思います。例えば「Cyclin-Dependent Kinase 4」は、SYMBOL型で略して「CDK4」と表されます。SYMBOLは、Human Genome Organization (HUGO) Gene Nomenclature Committeeによって管理され、国際的な標準として広く採用されています。
他にも以下のような表記方法があることを抑えておきましょう。下記はすべてCDK4を表しています。
- SYMBOL: CDK4
- Entrez Gene ID: 1019
- RefSeq: NM_000075.3
- Ensembl Gene ID: ENSG00000135446
- UniProtKB/Swiss-Prot: P11802
- HGNC: 1773
- MGI: 88346
clusterprofilerを使ったエンリッチメント解析の実装方法
実際にエンリッチメント解析を行っていきます。Rを実行する環境としてRstudioを使いますので、準備できていない方はこちらの記事より環境構築をお願いします。
clusterprofilerを使ったエンリッチメント解析の最小限のコードは以下になります。geneListに入れる遺伝子名はこちらで用意しておきました。
library(clusterProfiler)
geneList <- c("UBL5","NDUFB8","CHMP2B","PRPF8","NOSTRIN","MFAP1","CWC22",
"PLCH2","PRPF31","ATP6AP1","DSC3","CLN5","CHDC2","PIP","ZNF473","DHX8",
"RAB5A","NUP98","NUPL1","HRK","SLC41A3","SNRPD3","SNRPD2","PCGF6","GSK3A",
"SLC2A2","ARCN1","ANGPTL3","COPB1","COPB2","STARD5","CYB5R4","DMD","FUS",
"COPS6","NOS3","SFXN2","CNTN5","IQSEC1","CRADD","STAC3","COPZ1","SULT1C4",
"EFTUD2","SLC35A1","B4GALT2","THRSP","NHP2L1","ZNF224","NXF1","PDLIM3","SAMM50",
"MTFR1","SART1","PCDHGA1","GINS2","MPG","GJA3","UBE2A","ESAM","CLK3","EIF4A2",
"NUTF2","INMT","SCYL3","XIAP","TRIM28","MPZL1","LY6G6C","RBM14","TPRX1",
"ATP6V0B","NUP107","ABCD1","TGS1","RPS4X","CRNKL1","YTHDC1","PPAN","PRRT1",
"KRTCAP2","GDPD5","ZNF132","WDR18","ATP5F1","AMOTL1","RPS16","CCDC74B","CCDC74A",
"SF3B1","SF3B3","SF3B2","MYOCD","MLKL","PRSS8","CACTIN","EIF2S1","ZNF16",
"PGD","SRP54","AQR","DYNC1I1","DCLRE1B","CALCOCO2","EVC2","LRP1B","ZNF552",
"COPG1","EPRS","COPA","ATP6V1G1","PIEZO1","FCGR2A","CPLX1","SNRPB","BACE1",
"ZNF154","RAB29","ATP6V0E2","AHCY","SLC1A3")
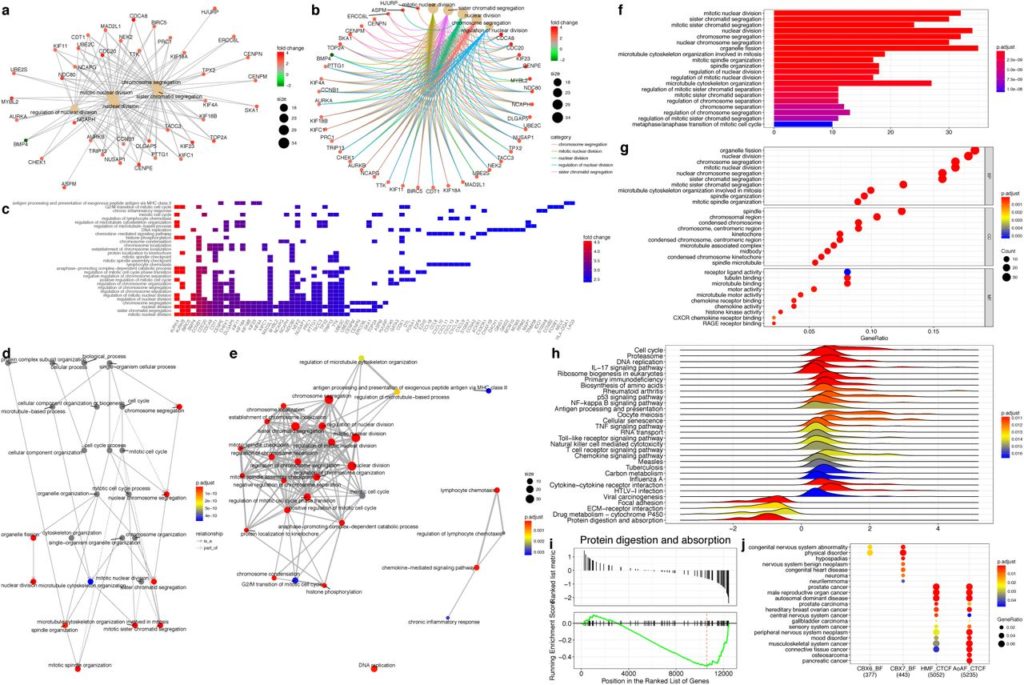
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
dotplot(result)実際に上記のコードを実行してみると、このような図が得られるかと思います。デフォルトではGeneRatio(クラスター内で解析対象のIDが占める割合)で図が作られます。
コードの内容を解説します。
library(clusterProfiler)でClusterProfilerパッケージを読み込んでいます。geneList <- c("UBL5","NDUFB8","CHMP2B"~~)で分析する遺伝子のリストを作成しています。result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL"):enrichGO関数を使って、遺伝子リストに基づくGOエンリッチメント解析を実行しています。OrgDbは、使用する生物種のデータベース(ここではヒト)を示しています。keyTypeは、遺伝子名のタイプ(ここではSYMBOL名)を指定しています。dotplot(result):dotplot関数を使って、エンリッチメント解析の結果を表示
実際に自分の遺伝子リストでやりたいときは、GeneList にはいる遺伝子名を変更してください。
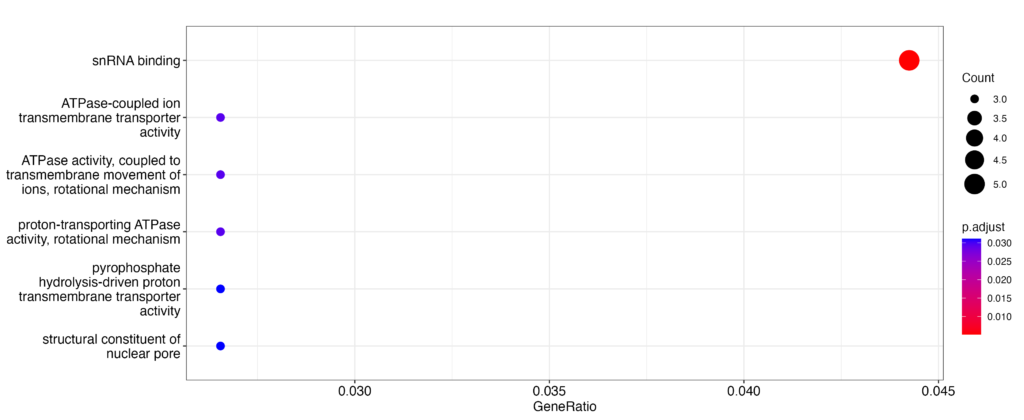
dotplotでxを指定してあげると、横軸を変更することが出来ます。試しにp.adjust(複数の検定を実行した場合に、p値を補正した値)に変更してみましょう
dotplot(result, x="p.adjust")結果は以下のようなイメージが取得できます。
他にも以下の値を用いて図の横軸を変更できます。色々と変えて試してみてください。
- ID: クラスターに属する解析対象のID (例えば、遺伝子IDやGO term IDなど)
- Description: 解析対象のIDの説明
- BgRatio: 全体の背景となる集合で解析対象のIDが占める割合
- pvalue: 解析対象のIDがクラスター内で偶然に集まっているかどうかを判断するための統計量のp値
- qvalue: False discovery rate (FDR) を制御するために用いられる統計量のq値
- geneID: クラスター内で解析対象のIDに対応する遺伝子名
- Count: クラスター内で解析対象のIDに対応する遺伝子の数
clusterprofilerでのその他の可視化方法
clusterprofilerを用いると他にも様々な可視化をすることが出来ます。色々な可視化を試してみて自分の解析結果の解釈に役立ちそうな図を探してみてください。
barplot
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
barplot(result)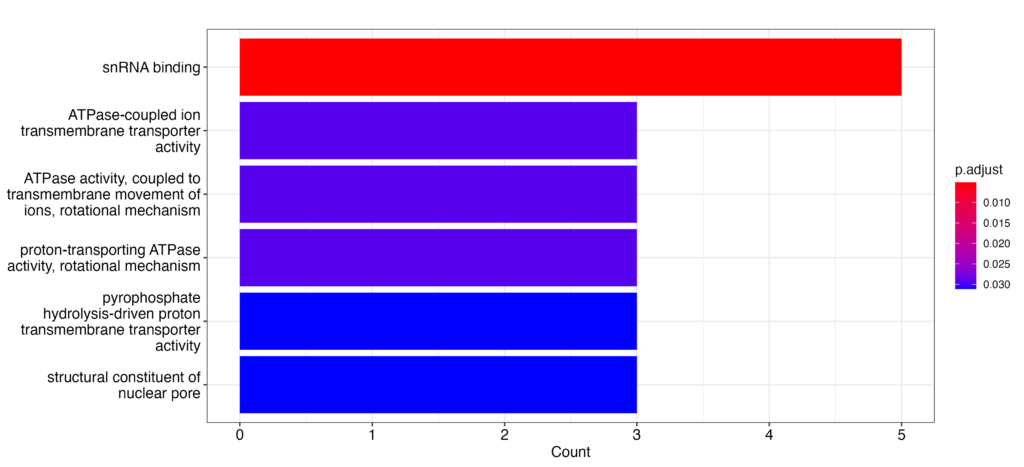
ggupset
BiocManager::install("ggupset")
library(enrichplot)
library(ggupset)
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
upsetplot(result)
emapplot
Gene OntologyやKEGG Pathwayなどの遺伝子オントロジー解析の結果を使った場合、エンリッチメントマップとして可視化することができます。重複する遺伝子セット間を結ぶネットワーク図を書くことが出来ます。
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
result2 <- pairwise_termsim(result)
emapplot(result2)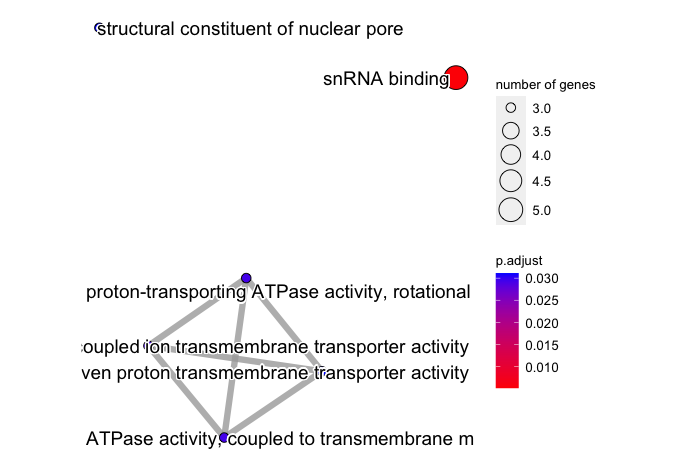
emapplotをするにはpairwise_termsim関数によって、与えられた2つ以上の用語集合の間の類似性を計算する必要がありますのでご注意ください。
pairwise_termsim関数は、共通のアノテーション項目を持つ2つ以上のリストを取り、そのアノテーション項目の共通性に基づいて、各リストのすべてのペアの類似性スコアを計算します。
goplot
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
goplot(result)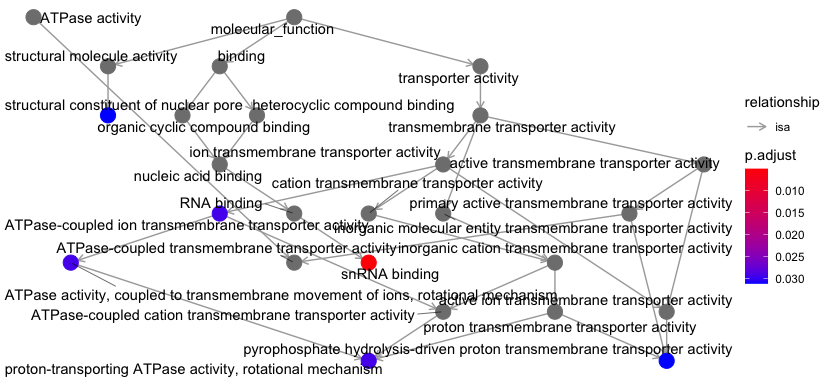
cnetplot
result <- enrichGO(geneList, OrgDb = "org.Hs.eg.db", keyType = "SYMBOL")
cnetplot(result)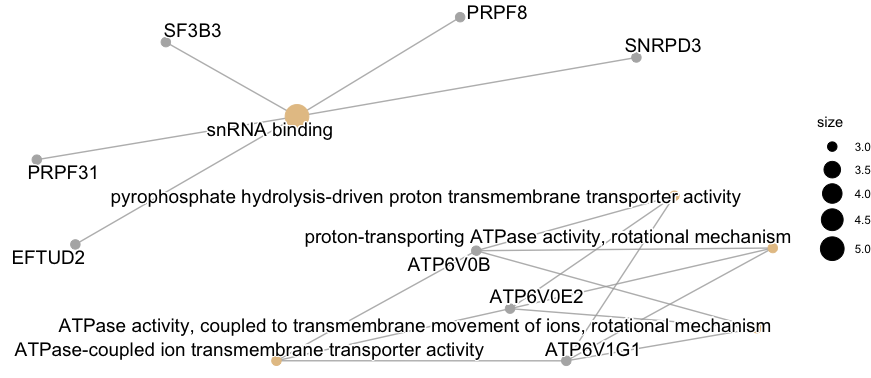
最後に
R言語のパッケージであるclusterprofilerを用いてエンリッチメント解析をする方法について解説しました。clusterprofilerをうまく使えば、特定のバイオマーカーや遺伝子セットが指定された生物学的なパスウェイや生物学的なプロセスにどの程度高確率で関連しているかを求めることが可能です。また豊富な可視化手段があり、エンリッチメント解析の結果を視覚的に表現することができますのでぜひともチャレンジしてみてください。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

