
single cell RNA-seq(scRNA-seq)に挑戦してみたい方はいるのではないでしょうか。しかしながら、scRNA-seqの計測機器はもっていないし、そもそも解析のやり方がわからない方がほとんどだと思います。
この記事では公共データからscRNA-seqデータを探してくる方法を説明します。
この記事を読むことで自分の研究に関連したscRNA-seqデータを公共データから見つけることができます。少し前置きが長いですが、全て大事な前提なのでしっかり理解していきましょう。
*本記事で扱っているscRNA-seq解析は、scRNA-seqデータを用いたデータ解析を指しており、サンプル調製の話ではないことにご注意ください。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
¥3,600 → ¥1,800 今なら50%OFF!!
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
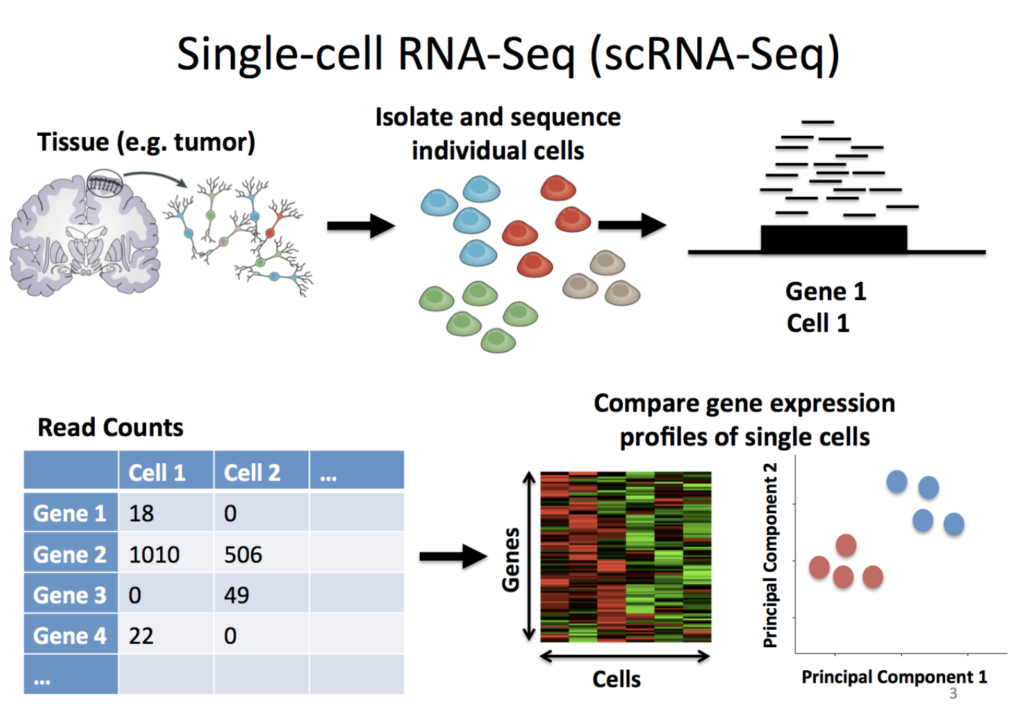
scRNA-seqとは?
scRNA-seq (single-cell RNA sequencing)は、個々の細胞内の遺伝子発現パターンを研究するために使用される方法です。これにより、細胞集団内の個々の細胞での遺伝子発現を分析することができます。細胞の識別、細胞のライフサイクル、細胞の発生などに用いられることが多い手法です。
Stephanie Hicks
Welcome to the World of Single-Cell RNA-Sequencing
https://speakerdeck.com/stephaniehicks/welcome-to-the-world-of-single-cell-rna-sequencing?slide=3
scRNA-seq解析をするときの言語はRとPythonどちらを選ぶか
Single cell RNA-seq解析には、PythonとRの両方のパッケージがあります。どちらを使うべきかは、それぞれに長所と短所があり、使用するにあたってのニーズや経験によって異なります。
■Python:ScanpyやCellRangerなど
Scanpy → 一般的なsingle cell RNA-seq解析に適している
CellRanger → 10x Genomics社のフォーマットに特化した分析に適している
■R:SeuratやMonocleなど
Seurat → 一般的なsingle cell RNA-seq解析に適している
Monocle→時系列データを扱う場合に適している
基本的にはどちらでもよく、自分が慣れている言語の方を選べば良いと思います。
PythonならScanpy、RならSeuratでライブラリ解析を行うという認識で良いと思います。
scRNA-seqデータベースについて
scRNA-seqデータはいくつかの公共データベースにも登録されています。
これらのデータベースにアクセスすることで、scRNA-seqデータを探すことができます。
- NCBI GEO – NCBIによって運営されている。バルクRNA-seqだけでなくscRNA-seqデータも提供
- Single Cell Expression Atlas (SCEA) – EMBL-EBIによって運営されているscRNA-seqデータベース
- Single Cell Portal(SCP)– The Broad Institute of MIT and Harvardが運営するscRNA-seqデータベース。2023年1月末時点で、507の研究課題と 29,614,655 cellsの発現量データを収録
- The Human Cell Atlas (HCA) – ヒト由来細胞のscRNA-seqデータを収録
- The Mouse Cell Atlas (MCA) - マウス由来細胞のscRNA-seqデータを収録
- SCPortalen– 理研が運用するマウスとヒトが中心のscRNA-seqデータベース
10x Genomics社のフォーマット
10x Genomics社は、シングルセルRNAシーケンシング技術を提供する企業で、低コストで大量の細胞を分析することができるため、研究者に広く普及しています。
10x Genomics社のものは独自のフォーマットで作成されています。10XGenome社の独自フォーマットを解析するために、cellrangerという特別なライブラリを提供していますが、こちらはSeuratでも解析可能ですので、今回はSeuratで解析します。
10x Genomics社独自フォーマットのデータは以下のようなファイル構成となっております。
$ tree filtered_feature_bc_matrix
filtered_feature_bc_matrix
├── barcodes.tsv.gz
├── features.tsv.gz (またはgenes.tsv.gz)
└── matrix.mtx.gz今回の記事ではこのようなファイルを探して、実際にクオリティチェックの解析までしてみようと思います。
scRNA-seqデータの探し方
大変前置きが長くなりましたが、scRNA-seqデータを公共データから探して、クオリティチェックの解析までしてみたいと思います。
クオリティチェックではSeuratを使いますので、こちらの記事を読んでRstudioやSeuratの事前知識があると良いです。
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(前編)【Seurat】
では実際にscRNA-seqデータを探してみましょう。今回はNCBI GEOからデータを探してみたいと思います。下記URLにアクセスしてください。
https://www.ncbi.nlm.nih.gov/geo/
右上の検索ボックスに検索ワードを打ち込みます。検索ワードは以下のように設定します。
"[検索したいワード]" and "single cell RNA-seq"今回は検索ワードは”lung cancer” and “single cell RNA-seq”としてください。
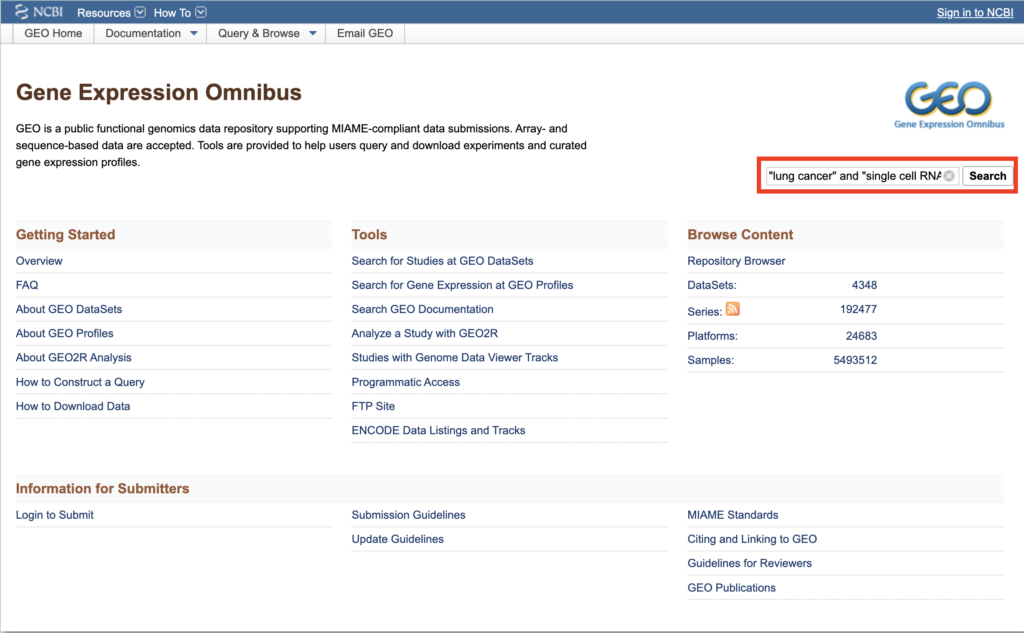
左サイドバーからcustomizeをクリックし「Expression profiling by high througput sequencing」にチェックを入れます。
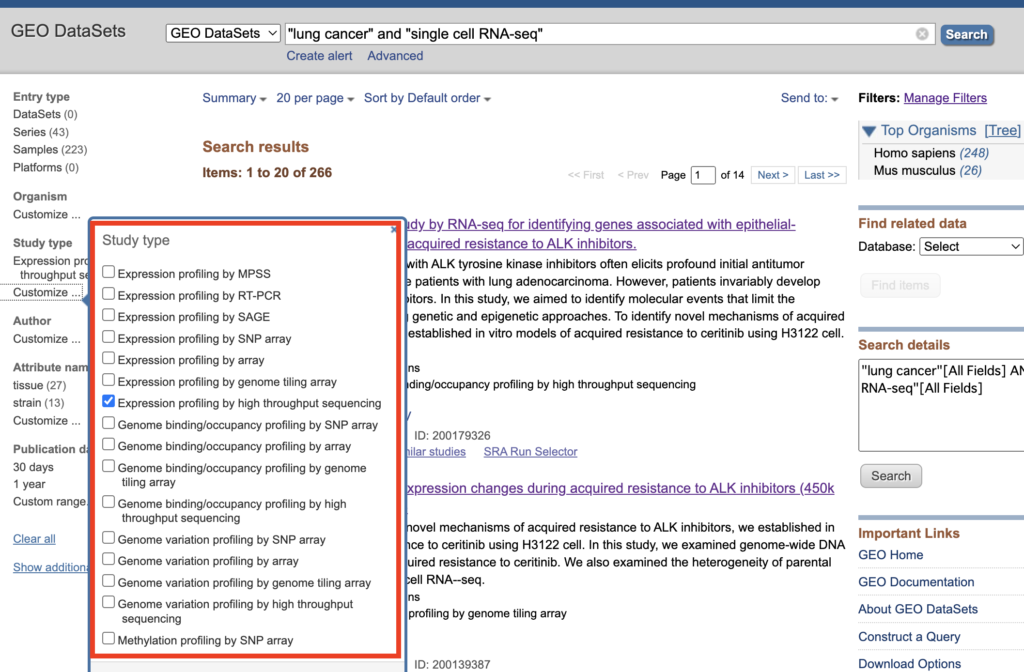
チェックしたあとに、「Expression profiling by high througput sequencing」が表示されますのでクリックをしてください。
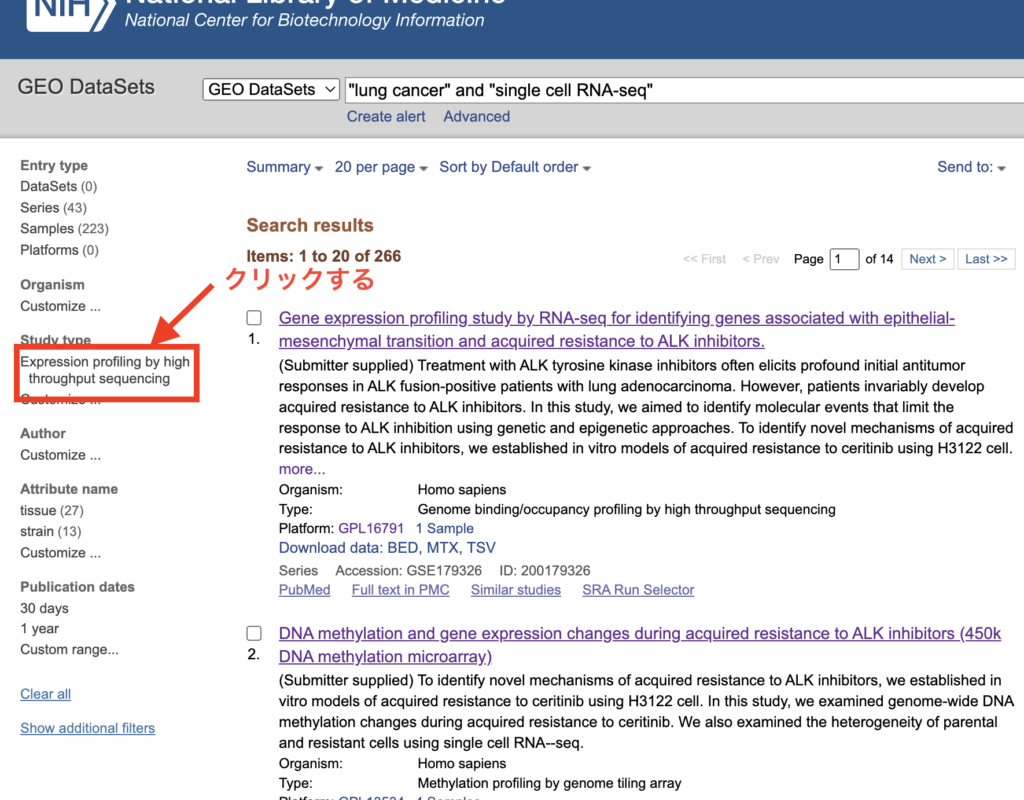
これで準備完了です。次に、scRNA-seq解析ができるサンプルを探してみます。
試しにトップにあるDNA methylation and gene expression changes during acquired resistance to ALK inhibitors (scRNA-seq)を見てみましょう。
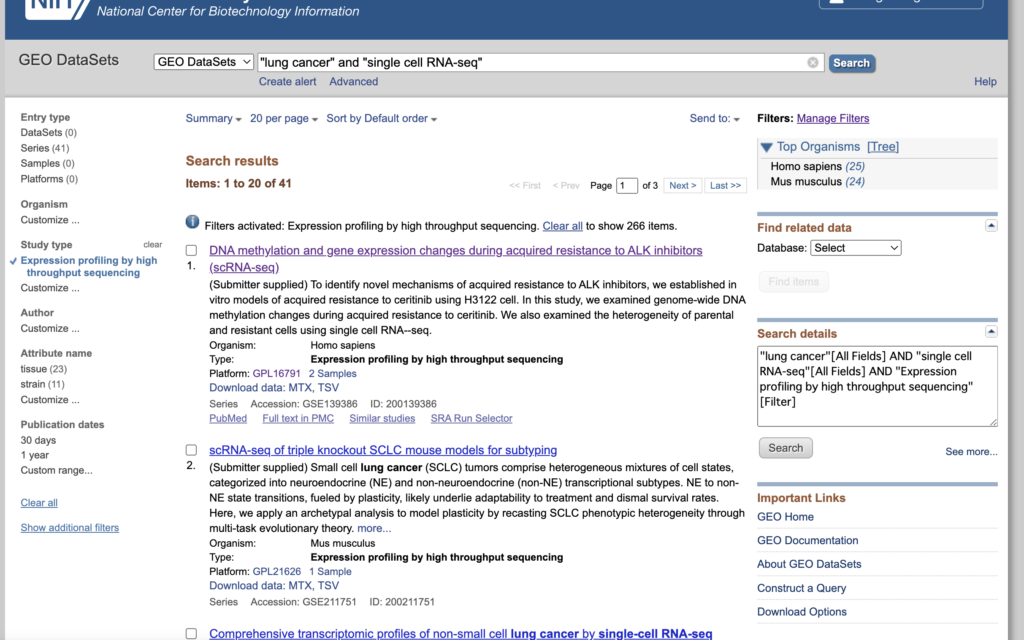
下にスクロールすると、Supplementary fileの情報が書かれています。
「custom」をクリックすると、中身のファイルが閲覧できます。
このサンプルでは、barcodes, features, matrixという10X genomics社のフォーマットファイルが揃っており、今回の解析で使うことができます。
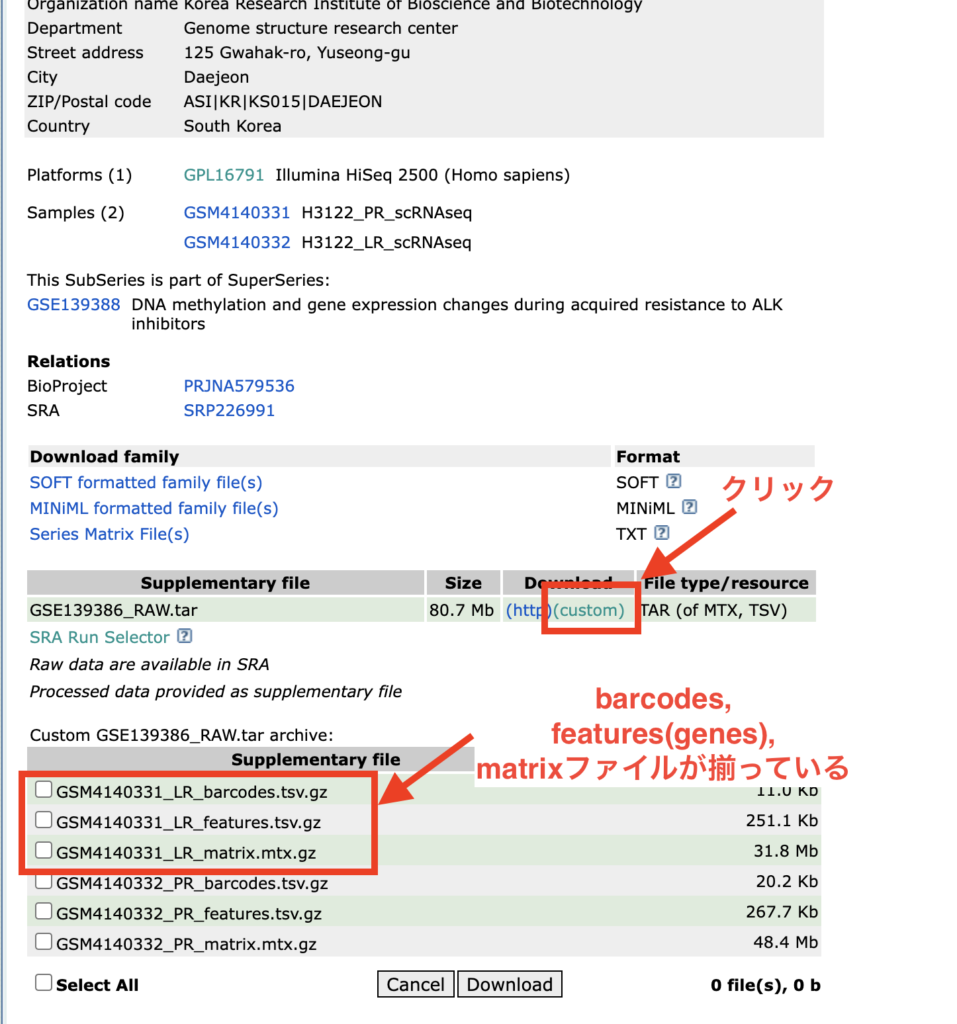
次に今回の解析が使えないパターンを紹介します。
検索結果にあったComprehensive transcriptomic profiles of non-small cell lung cancer by single-cell RNA-seqを見てみましょう。RAW.tarファイルの中を見るとこのようにtxt.gzが入っています。
このようなパターンは今回の解析には用いれないので避けてください。また別の機会に紹介します。
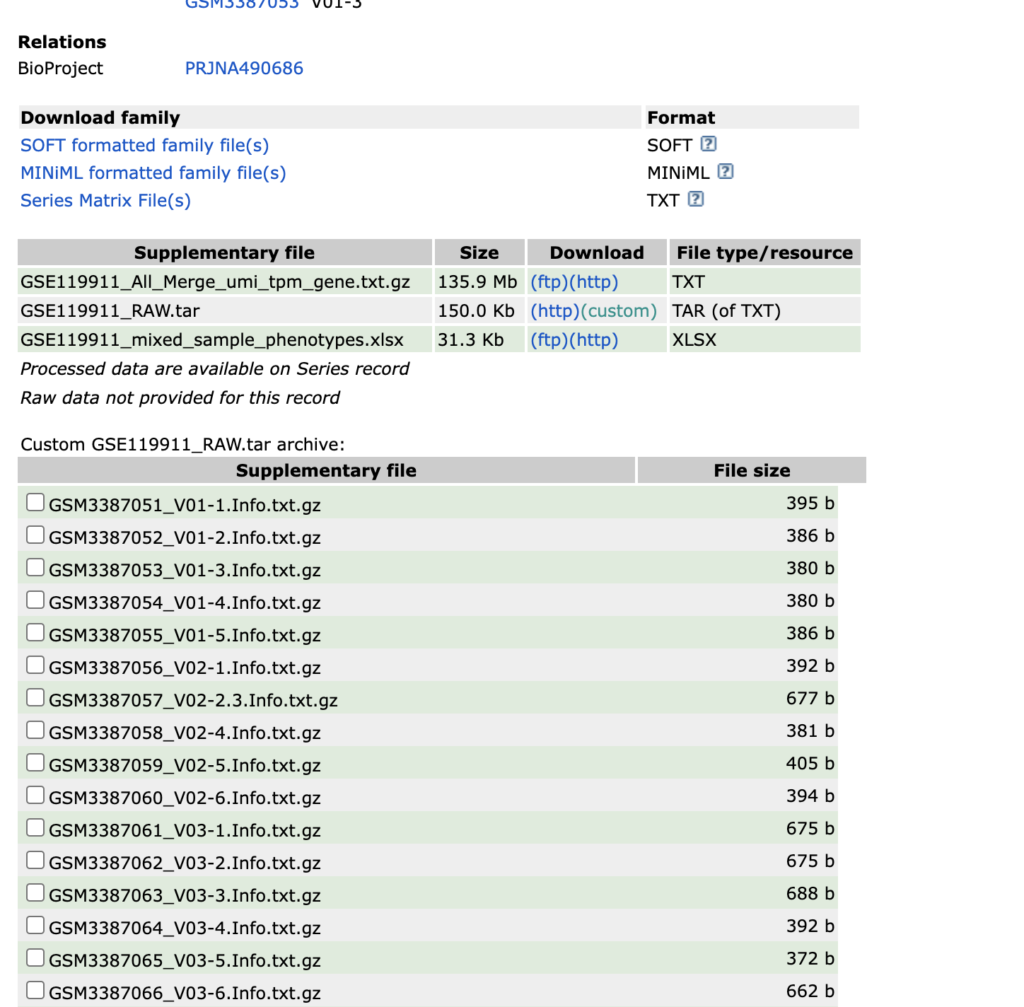
それではDNA methylation and gene expression changes during acquired resistance to ALK inhibitors (scRNA-seq)を実際に解析してみましょう。
ファイルにチェックボックスにチェックを入れてDownloadをクリックするとtarファイルとしてダウンロードされますので、デスクトップに持っていき解凍してください。
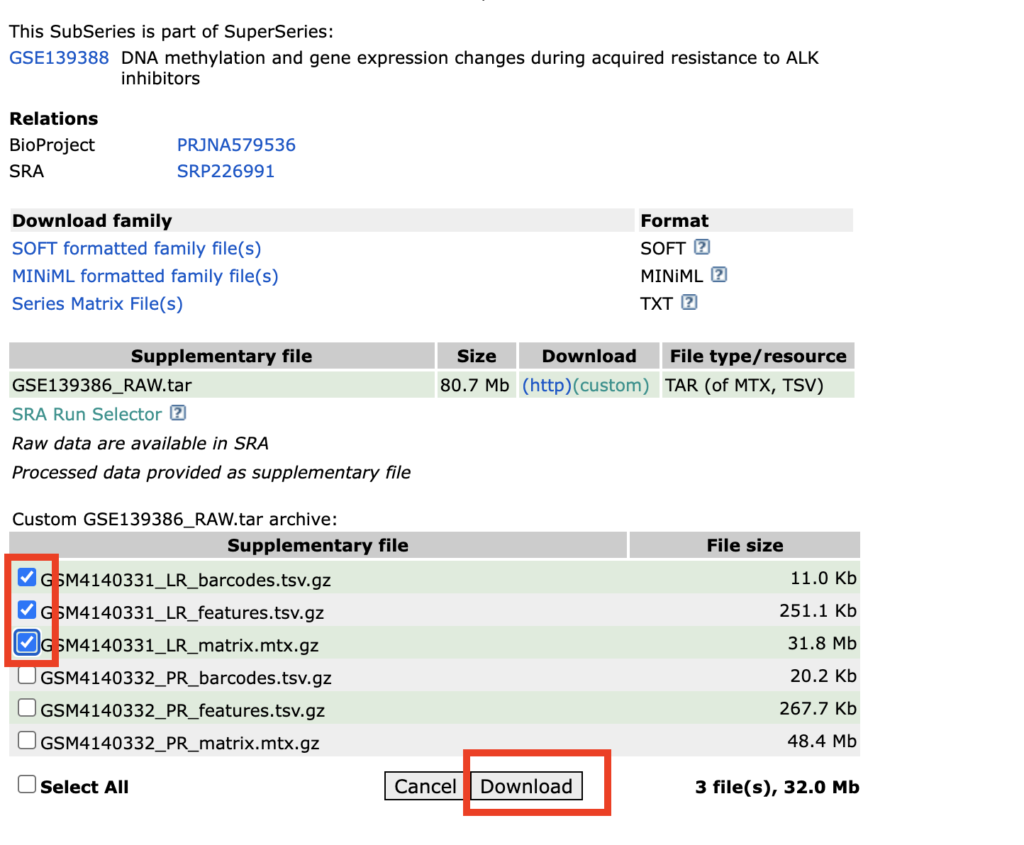
ダウンロードしたサンプルの中身はこの様になっています。
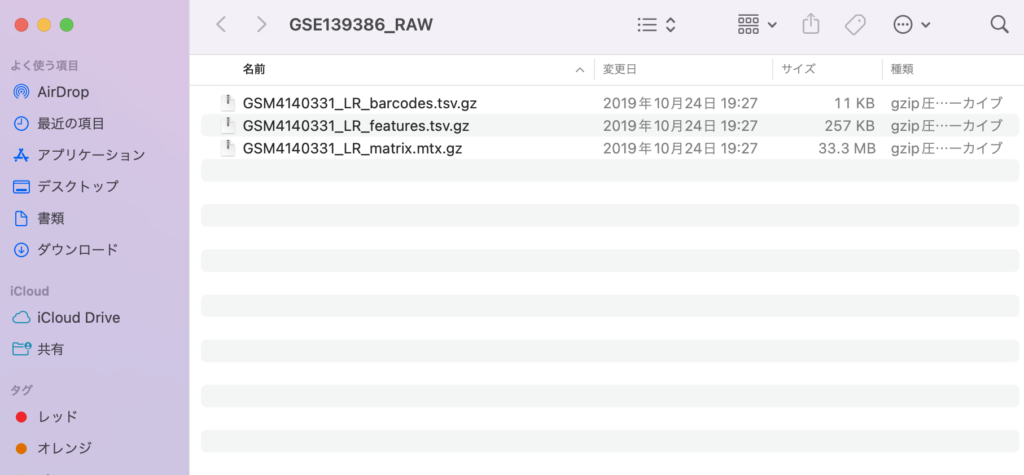
次にファイル名をbarcodes.tsv.gz, features.tsv.gz, matrix.mtx.gzにそれぞれ変更してください。
この操作をしないとSeuratで読み込むことができません。
ちなみにgz形式のままで大丈夫なので、解凍はしなくて良いです。
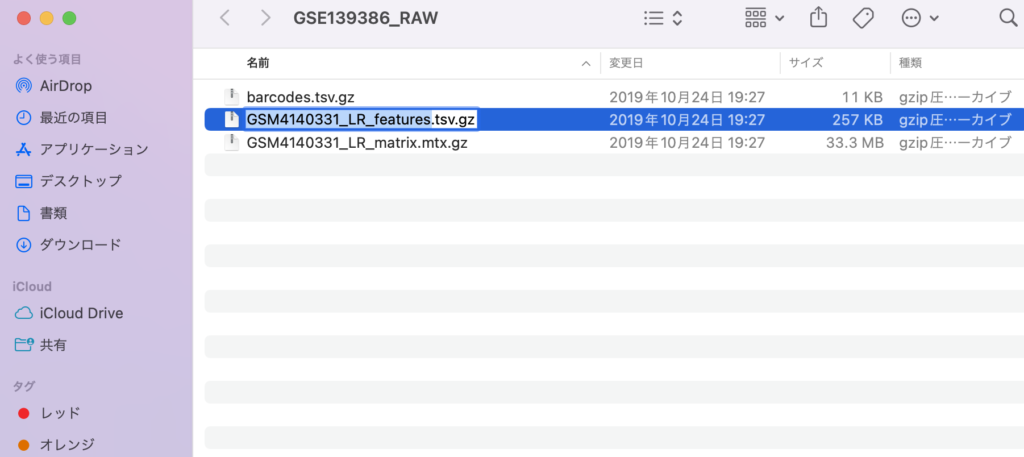
以上で準備は完了です。
次にSeuratでファイルを読み出してみようと思います。
Seuratを使うときはRstudioをつかいますので、Rstudioの使い方がわからない方はこちらの記事を参考に準備をお願いします。
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(前編)【Seurat】
Rstudioの右下画面でデスクトップまで進み、
- ダウンロードしてきたファイルにチェック
- Moreをクリック
- Set As Working Directory
を順に選びます。この作業を間違うと後のコードが一切通らなくなるのでお気をつけください。
必要なコードは以下の通りです。まずはライブラリーを読み出していきます。
install.packages("dplyr")
install.packages("Seurat")
install.packages("patchwork")
library(dplyr)
library(Seurat)
library(patchwork)以下のコードでサンプルを読み出して、クオリティチェックのためのバイオリンプロットを表示してみましょう。
# サンプルを読み出す
pbmc_v2.data <- Read10X(data.dir = "./GSE139386_RAW/")
# バイオリンプロットを表示する
pbmc_v2 <- CreateSeuratObject(counts = pbmc_v2.data, project = "pbmc_v23k", min.cells = 3, min.features = 200)
pbmc_v2[["percent.mt"]] <- PercentageFeatureSet(pbmc_v2, pattern = "^MT-")
VlnPlot(pbmc_v2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)以下のようなバイオリンプロットが表示できたら成功です!
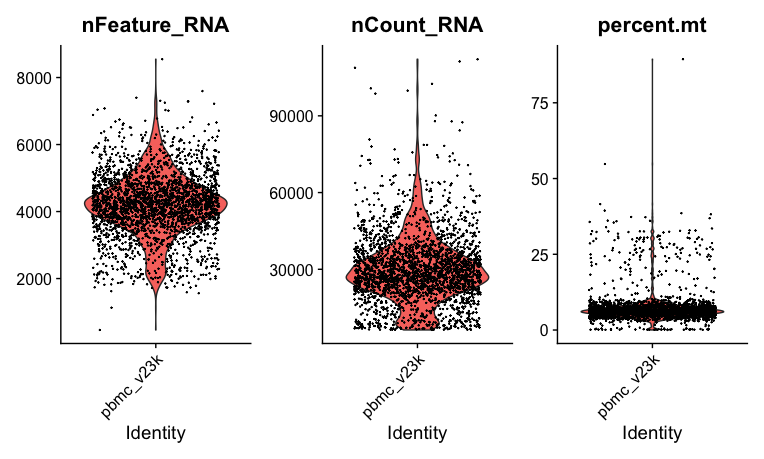
残りの解析は、Seuratのハンズオン記事を書いております。こちらを参考にscRNA-seqのいろいろな可視化にチャレンジしてみてください!
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(前編)【Seurat】
【scRNA-seq】Seuratを用いてscRNA-seq解析を始める方法(後編)【Seurat】
最後に
いかがだったでしょうか。自分の研究に関係のあるsingle cell RNA-seqデータを公共データから探すイメージがついたなら幸いです。ぜひ自分の好きなデータを見つけて再解析してみましょう。

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

