
本記事はAI創薬の一つである機械学習を用いたin silico screeningについて書かれた記事です。第5章まであり、すべての内容が理解できると、目的の標的にあった薬物候補化合物を発見することができます。こちらの記事は第5章で第4章で作った学習モデルを使ってin silico screeningを行います!ぜひ皆さんもトライしてみて下さい!
macOS Ventura(13.2.1), Google colaboratory
今回は直ぐにpythonが実行できるようにGoogle colabを利用していきます。

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1500 今なら50%OFF!!
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1,500 今なら50%OFF!!
分子ドッキングやMDシミュレーションなど、
自宅でできるin silico創薬の解析方法を解説したものになります!
AI創薬とは?
AI創薬は、人工知能(AI)技術を利用して新しい薬物を発見、開発するプロセスです。AIは大量のデータを高速に処理し、薬物の候補を予測したり、薬物相互作用を評価したりします。また、AIは薬物の効果や安全性をシミュレートすることも可能で、臨床試験の前の段階でリスクを評価することができます。これにより、薬物開発のコストと時間を大幅に削減することが期待されています。AI創薬は、薬物開発の新しいパラダイムとして注目を集め、製薬企業や研究機関で積極的に研究、導入が進められています。また、バイオインフォマティクス、ケモインフォマティクス、機械学習、ディープラーニングなどの技術が組み合わされ、薬物開発のプロセスを革新しています。さらに、AI創薬は個人化医療の推進にも寄与し、患者にとって最適な治療法を提供する可能性を秘めています。
今回はAI創薬の中でも、in silico screeeningに焦点を当てて解説してきます。
以下の論文、記事をフォローしていきます。
NCBI – WWW Error Blocked Diagnostic
論文と公共データベースを使って無料で始めるAI創薬 – Qiita
この論文では新型コロナウイルスの標的タンパク質である3C-like プロテアーゼ(メインプロテアーゼ:Mpro)に対して、公共データベースにある化合物でスクリーニングを行い、最終的に41個のの化合物を薬物候補化合物としています。
AI創薬によるIn silico screeningの流れ
以下のような流れで行っていきます。全5章となります。
- 公共データベース(ChEMBL)からの機械学習の学習データを収集
- スクレイピングによる公共データベース(PDB)からの機械学習の学習データを収集
- 機械学習の学習データの整形
- 整形データを用いた予測モデルの作成
- 予測モデルによる候補化合物のin silico screening(本記事)
本記事はいよいよ第5章 予測プログラムによる候補化合物のin silico screeningになります。
この章では以下の順番で、in silico screeningを行っていきます。
- 化合物集団(化合物ライブラリ)をDrug Bankから取得
- in silico screening
Drug Bankからの対象データの出力
in silico screeningする対象のデータはDrug Bankからのデータになります。
Drug Bankの会員登録を行って、以下にアクセスしてください。
https://go.drugbank.com/releases/latest#structures
次にApprovedと書いてあるDrug Groupをダウンロードしてください。こちらはすでに違う病気に効く薬として承認された化合物ライブラリです。今回はこちらの化合物ライブラリをスクリーニングします。この化合物ライブラリは既に承認されているため、薬物の安全性が保証されています。ですので、もしこの中からコロナにも効く化合物が見つかれば、臨床試験をある程度簡易化することができ、薬の開発ステップを削減できます。このように既存の薬剤に対して、新しい作用を見つけることをドラックリポジショニングと言います。
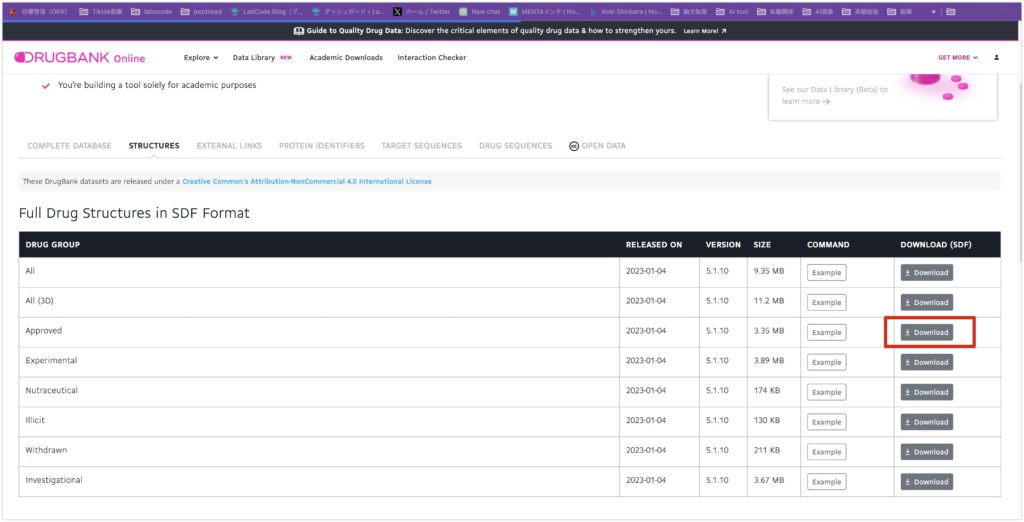
ダウンロードしたファイルをgoogle colabにアップロードしてください。
また第3章で作成したmerged_file_path.csv , 第4章で作成したoutput_model.pkl.gz をアップロードしておいてください。
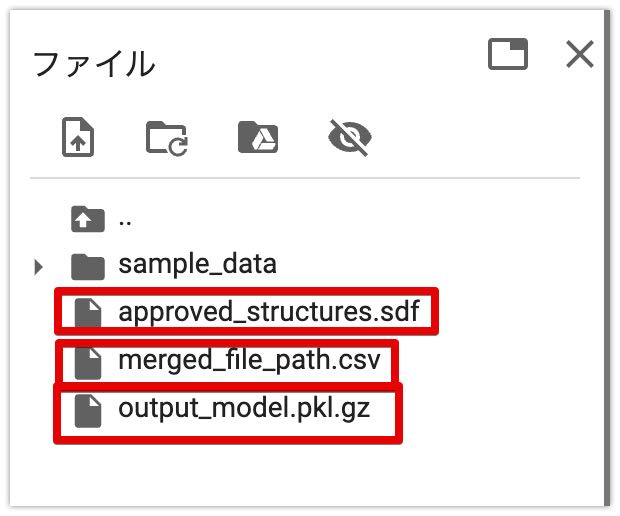
これにて準備は完了です。
Drug Bankデータの整形
まずDrug Bankデータを使用できるように整形していきます。
以下のコードを実行してください。ライブラリのインストールとインポート、ファイルパスの定義を行っています。
!pip install rdkit-pypi pandas
!pip install molvs
from rdkit import Chem
from rdkit.Chem import AllChem
from molvs.normalize import Normalizer, Normalization
from molvs.tautomer import TautomerCanonicalizer, TautomerEnumerator
from molvs.fragment import LargestFragmentChooser
from molvs.charge import Reionizer, Uncharger
import pandas as pd
import numpy as np
import gzip
import _pickle as cPickle
import csv
input_sdf = "/content/approved_structures.sdf"
train_data = "/content/merged_file_path.csv"
- ライブラリのインストール:
!pip install rdkit-pypi pandas: **rdkitという化学情報処理用のライブラリと、データ分析用のpandas**ライブラリをインストールします。!pip install molvs: **molvs**は化合物の標準化、タウトメリズムの処理、イオン状態の調整など、化学情報の前処理に使われるライブラリです。
- ライブラリのインポート:
from rdkit import Chemとfrom rdkit.Chem import AllChem: **rdkit**から化学構造の操作や変換に関連するモジュールをインポートします。from molvs.normalize import Normalizer, Normalization: 化合物の標準化を行うためのクラスとメソッドを**molvs**からインポートします。from molvs.tautomer import TautomerCanonicalizer, TautomerEnumerator: タウトメリズムのバリエーションを処理するためのクラスをインポートします。from molvs.fragment import LargestFragmentChooser: 複数のフラグメント(分子の部分)の中から最大のフラグメントを選択するためのクラスをインポートします。from molvs.charge import Reionizer, Uncharger: イオン化状態の調整に関するクラスをインポートします。import pandas as pd: **pandasライブラリをpd**という名前でインポートします。import numpy as np: 数値計算に広く使われる**numpyライブラリをnp**という名前でインポートします。import gzip: gzip形式のファイルを扱うためのライブラリをインポートします。import _pickle as cPickle: Pythonオブジェクトの保存や読み込みに使われる**pickle**ライブラリをインポートします。import csv: CSVファイルの読み書きに使われる標準ライブラリをインポートします。
- ファイルパスの定義:
input_sdf = "/content/approved_structures.sdf": SDF形式のファイル(化学構造データ)のパスを指定します。train_data = "/content/merged_file_path.csv": トレーニングデータを含むCSVファイルのパスを指定します。
次に以下を実行してください。
def normalize(smiles):
# Generate Mol
mol1 = Chem.MolFromSmiles(smiles)
# Uncharge
uncharger = Uncharger()
mol2 = uncharger.uncharge(mol1)
# LargestFragmentChooser
flagmentChooser = LargestFragmentChooser()
try:
mol3 = flagmentChooser(mol2)
except:
try:
mol3 = flagmentChooser(mol1)
except:
mol3 = mol1
# Sanitaize
Chem.SanitizeMol(mol3)
# Normalize
normalizer = Normalizer()
mol4 = normalizer.normalize(mol3)
#tautomerCanonicalizer = TautomerCanonicalizer()
#mol = tautomerCanonicalizer.canonicalize(mol)
return Chem.MolToSmiles(mol4)
このPython関数 normalize は、SMILES(Simplified Molecular Input Line Entry System)形式で表された化学構造を標準化するために使用されます。SMILESは化学構造を線形の文字列で表現する方法です。この関数は以下のステップで化合物を処理します:
- Molオブジェクトの生成:
mol1 = Chem.MolFromSmiles(smiles): 入力されたSMILES文字列からRDKitのMolオブジェクトを生成します。
- アンチャージ(イオン化状態の調整):
uncharger = Uncharger(): Unchargerオブジェクトを作成します。mol2 = uncharger.uncharge(mol1): Molオブジェクトのイオン化状態を調整します。
- 最大フラグメントの選択:
flagmentChooser = LargestFragmentChooser(): 最大フラグメントを選択するオブジェクトを作成します。mol3 = flagmentChooser(mol2): Molオブジェクトから最大のフラグメントを選択します。もしエラーが発生した場合は、元のMolオブジェクト(mol1)を使用して再試行します。
- サニタイズ(構造の検証と修正):
Chem.SanitizeMol(mol3): Molオブジェクトの構造を検証し、必要に応じて修正します。
- 標準化:
normalizer = Normalizer(): Normalizerオブジェクトを作成します。mol4 = normalizer.normalize(mol3): Molオブジェクトを標準化します。
- タウトメリズムの処理:
- この部分はコメントアウトされているため、実行されませんが、タウトメリズムのバリエーションを標準化するために使用されるコードです。
- SMILES形式への変換:
return Chem.MolToSmiles(mol4): 処理されたMolオブジェクトをSMILES形式に変換し、それを戻り値として返します。
この関数は、化合物のデータセットを扱う際に一貫した形式で化学構造を扱うために重要で、特に大規模な化学データベースや機械学習アプリケーションで有用です。
続いて、以下を実行してください。
# Drug Bankデータの読み込み、整形
data = []
sdf_sup = Chem.SDMolSupplier(input_sdf)
for i, mol in enumerate(sdf_sup):
if not mol:
continue
if mol.HasProp("DATABASE_ID"):
id = mol.GetProp("DATABASE_ID")
if mol.HasProp("GENERIC_NAME"):
name = mol.GetProp("GENERIC_NAME")
smiles = Chem.MolToSmiles(mol)
new_smiles = normalize(smiles)
data.append([id, name, new_smiles])
# pandas DataFrameにデータを変換
df = pd.DataFrame(data, columns=['id', 'name', 'smiles'])
# DataFrameをExcelファイルとして保存
df.to_csv('output.csv', index=False)
このコードは、SDファイルから化学構造データを読み込み、それを標準化して、PandasのDataFrameに変換し、最後にCSVファイルとして保存するプロセスを行っています。各ステップを詳しく説明します:
- データリストの初期化:
data = []: 空のリストを作成して、後で化合物データを格納します。
- SDファイルの読み込み:
sdf_sup = Chem.SDMolSupplier(input_sdf): 指定されたSDファイルから化合物データを読み込むためのサプライヤー(SDMolSupplier)を作成します。
- データの読み込みと処理:
for i, mol in enumerate(sdf_sup): ファイル内の各化合物に対してループを行います。if not mol: continue: 化合物が無効な場合(例えば、読み込みエラーが発生した場合)、その化合物をスキップします。if mol.HasProp("DATABASE_ID"): 化合物にデータベースIDがある場合のみ、次の処理を行います。id = mol.GetProp("DATABASE_ID"): 化合物のデータベースIDを取得します。if mol.HasProp("GENERIC_NAME"): 化合物にジェネリック名がある場合、それを取得します。name = mol.GetProp("GENERIC_NAME"): 化合物のジェネリック名を取得します。smiles = Chem.MolToSmiles(mol): 化合物をSMILES形式に変換します。new_smiles = normalize(smiles): SMILES形式の化合物を標準化する**normalize**関数を呼び出します。data.append([id, name, new_smiles]): 標準化されたSMILES、ID、名前をデータリストに追加します。
- Pandas DataFrameの作成:
df = pd.DataFrame(data, columns=['id', 'name', 'smiles']): 収集したデータをPandasのDataFrameに変換します。列名は「id」、「name」、および「smiles」です。
- CSVファイルとして保存:
df.to_csv('output.csv', index=False): DataFrameを**output.csvという名前のCSVファイルに変換して保存します。index=False**は、DataFrameのインデックスをCSVファイルに含めないことを指定します。
このプロセスを通じて、化学データベースからの情報を効率的に収集・整理し、さらなる分析や共有のためにアクセスしやすい形式に変換することができます。
続いて以下を実行してください。
# 予測データに含まれる学習データの削除
# CSVファイルの読み込み
input_predict = "/content/output.csv"
traindata_df = pd.read_csv(train_data)
input_predict_df = pd.read_csv(input_predict)
# 'canonical_smiles'の値でフィルタリング
filtered_input_predict_df = input_predict_df[~input_predict_df['smiles'].isin(traindata_df['canonical_smiles'])]
# 結果の保存
predict_data = "/content/predictdata.csv"
filtered_input_predict_df.to_csv(predict_data, index=False)
このコードの目的は、予測データセットから学習データセットに含まれる化合物を削除することです。これは、モデルの評価時にテストデータが学習データに含まれていないことを確認するために重要です。コードの各ステップを詳しく説明します:
- CSVファイルの読み込み:
input_predict = "/content/output.csv": 予測データセットのファイルパスを指定します。traindata_df = pd.read_csv(train_data): 学習データセットをPandasのDataFrameとして読み込みます。input_predict_df = pd.read_csv(input_predict): 予測データセットをPandasのDataFrameとして読み込みます。
- フィルタリング処理:
filtered_input_predict_df = input_predict_df[~input_predict_df['smiles'].isin(traindata_df['canonical_smiles'])]: 予測データセットから、学習データセットの**canonical_smiles**列に含まれるSMILESを持つ化合物を除外します。- **
~は否定を意味し、isin**関数は指定された値が列に含まれているかどうかをチェックします。これにより、学習データに含まれていない化合物のみを選択します。
- **
- 結果の保存:
predict_data = "/content/predictdata.csv": フィルタリングされたデータを保存するファイルパスを指定します。filtered_input_predict_df.to_csv(predict_data, index=False): フィルタリングされたデータをCSVファイルとして保存します。**index=False**は、DataFrameのインデックスをCSVファイルに含めないことを指定します。
このプロセスにより、モデルの学習と評価をより効果的に行うために、学習データと予測データの間での重複を排除することができます。これは、特に機械学習モデルの一般化能力を評価する際に重要です。
続いて、以下のコードを実行してください。
#予測データの読み込み
smiles_list = list()
datas = []
predict_data_fp = "/content/predictdatafp.csv"
# 予測データの読み込み
with open(input_predict, mode='r', encoding='utf-8') as f:
reader = csv.DictReader(f)
for row in reader:
print(row["id"])
smiles = row["smiles"]
new_smiles = normalize(smiles)
smiles_list.append((row["id"], new_smiles))
mol = Chem.MolFromSmiles(new_smiles)
fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius=3, nBits=2048, useFeatures=False, useChirality=False)
fp = pd.Series(np.asarray(fp)).values
data = []
data.append(row["id"])
data.extend(fp)
datas.append(data)
columns = list()
columns.append("id")
columns.extend(["Bit_" + str(i+1) for i in range(2048)])
df = pd.DataFrame(data=datas, columns=columns)
df.set_index("id", inplace=True, drop=True)
#一旦保存
df.to_csv(predict_data_fp)
このコードは、予測データを読み込んで処理し、化学構造の特徴を表すフィンガープリント(指紋)を生成し、最後に結果をCSVファイルとして保存するプロセスを実装しています。具体的な手順は以下のとおりです:
- リストの初期化:
smiles_list = list(): SMILES文字列とIDのペアを格納するための空のリスト。datas = []: 最終的なデータセットを格納するための空のリスト。
- 予測データファイルパスの設定:
predict_data_fp = "/content/predictdatafp.csv": 処理後のデータを保存するファイルのパス。
- 予測データの読み込みと処理:
with open(input_predict, mode='r', encoding='utf-8') as f: 予測データファイルを読み込みます。reader = csv.DictReader(f): CSVファイルを辞書形式で読み込むためのリーダーを作成します。for row in reader: 各行に対してループを実行します。smiles = row["smiles"]: 各行からSMILES文字列を取得します。new_smiles = normalize(smiles): SMILES文字列を標準化します。smiles_list.append((row["id"], new_smiles)): 標準化されたSMILESとIDのペアをリストに追加します。mol = Chem.MolFromSmiles(new_smiles): SMILESからMolオブジェクトを生成します。fp = AllChem.GetMorganFingerprintAsBitVect(...): Molオブジェクトからモルガンフィンガープリントを生成します。fp = pd.Series(np.asarray(fp)).values: フィンガープリントをNumPy配列に変換し、Pandasのシリーズにして値を取得します。data = [row["id"]] + list(fp): IDとフィンガープリントを結合してデータリストに追加します。
- DataFrameの作成と保存:
columns = ["id"] + ["Bit_" + str(i+1) for i in range(2048)]: 列名を作成します。df = pd.DataFrame(data=datas, columns=columns): データと列名を使ってDataFrameを作成します。df.set_index("id", inplace=True, drop=True): ‘id’列をインデックスとして設定します。df.to_csv(predict_data_fp): DataFrameをCSVファイルとして保存します。
このコードは、化学構造の特徴を捉えるフィンガープリントを生成し、それを含むデータセットを機械学習モデルの予測入力として利用するためのものです。モルガンフィンガープリントは、各化合物の化学的特性を表すバイナリベクトルで、機械学習アルゴリズムが理解できる形式で化合物を表現します。
これにて、Drug data Bankの整形は終了です!
In silico screening
いよいよ待ちに待ったin silico screeningを行っていきます。
まず以下を実行してください。
# モデルの読み込み
X_vs = df.iloc[:, 0:]
input_model = "/content/output_model.pkl.gz"
with gzip.GzipFile(input_model, 'r') as f:
model = cPickle.load(f)
このコードのセクションは、保存された機械学習モデルを読み込むためのものです。具体的なステップは以下の通りです:
- モデル入力データの準備:
X_vs = df.iloc[:, 0:]: 作成されたDataFramedfから、すべての行と列を選択してモデルの入力データ (X_vs) として用います。ここでは、行の全範囲と列の全範囲を選択しています。
- モデルファイルパスの設定:
input_model = "/content/output_model.pkl.gz": 読み込むべき機械学習モデルのファイルパスを指定します。このファイルはgzipで圧縮されたpickleファイルとして保存されていると想定されます。
- モデルの読み込み:
with gzip.GzipFile(input_model, 'r') as f: gzip形式のファイルを読み込むためにgzip.GzipFileを使用します。ここで'r'は読み込みモードを意味します。model = cPickle.load(f): pickleファイルから機械学習モデルを読み込みます。cPickle.load関数は、pickle化されたオブジェクト(この場合は機械学習モデル)をPythonオブジェクトに変換します。
このコードは、以前に訓練されて保存された機械学習モデルをロードし、それを使用して新しいデータセット上で予測を行うために使われます。モデルの読み込みには、保存時に使用された同じライブラリとモデルの構造が必要です。圧縮されたpickleファイルを使用することで、ファイルサイズを小さく保ち、読み込みや保存のプロセスを効率化できます。
続いて、次のコードを実行してください。
# モデルの確信度を0.7以上に設定し、予測
ad_threshold = 0.70
y_pred = model.predict(X_vs)
confidence = model.predict_proba(X_vs)
confidence = np.amax(confidence, axis=1).round(2)
ad = confidence >= ad_threshold
pred = pd.DataFrame({'Prediction': y_pred, 'AD': ad, 'Confidence': confidence}, index=None)
pred.AD[pred.AD == False] = np.nan
pred.AD[pred.AD == True] = pred.Prediction.astype(int)
pred_ad = pred.dropna().astype(int)
coverage_ad = len(pred_ad) * 100 / len(pred)
print('VS pred: %s' % pred.Prediction)
print('VS pred AD: %s' % pred_ad.Prediction)
print('Coverage of AD: %.2f%%' % coverage_ad)
このコードは、予測された結果とそれに伴う確信度(confidence)を分析し、確信度が設定された閾値(0.7)以上の予測のみを抽出し、さらにそれらの予測が全体に占める割合(カバレッジ)を計算しています。以下に各ステップを説明します:
- 確信度の閾値設定と予測の実行:
y_pred = model.predict(X_vs): モデルによる予測結果(クラスラベル)。confidence = model.predict_proba(X_vs): 各予測に対するモデルの確信度(クラス毎の確率)。confidence = np.amax(confidence, axis=1).round(2): 各サンプルに対して最高の確信度を選び、小数点以下2桁で丸めます。ad = confidence >= ad_threshold: 確信度が設定された閾値0.7以上の場合にTrueとなるブール値の配列。
- 予測結果のDataFrameの作成:
pred = pd.DataFrame(...): 予測結果、確信度が閾値以上かどうかの情報(AD)、および確信度自体を含むDataFrameを作成します。
- 確信度が閾値未満の予測の除外:
pred.AD[pred.AD == False] = np.nan: 確信度が閾値未満の場合、その行のAD値をNaNに置き換えます。pred.AD[pred.AD == True] = pred.Prediction.astype(int): 確信度が閾値以上の場合、その行のAD値を予測値に置き換えます。
- NaNを除外して確信度が高い予測のみを保持:
pred_ad = pred.dropna().astype(int): NaNを含む行を除外し、残りのデータを整数型に変換します。
- 確信度が高い予測のカバレッジ(割合)の計算:
coverage_ad = len(pred_ad) * 100 / len(pred): 確信度が高い予測の数を全予測の数で割り、カバレッジのパーセンテージを計算します。
- 結果の表示:
print('VS pred: %s' % pred.Prediction): 全予測の表示。print('VS pred AD: %s' % pred_ad.Prediction): 確信度が高い予測の表示。print('Coverage of AD: %.2f%%' % coverage_ad): 確信度が高い予測のカバレッジの表示。
このコードは、モデルの予測結果の信頼性を評価し、確信度が高い予測のみを取り出して、その予測の品質とカバレッジを評価するために使われます。
最後に以下のコードを実行してください。
prediction_report="/content/prediction_report.csv"
pred.index = X_vs.index
predictions = pd.concat([pred], axis=1)
for col in ['Prediction', 'AD']:
predictions[col].replace(0, 'Inactive', inplace=True)
predictions[col].replace(1, 'Active', inplace=True)
print(predictions.head())
predictions.to_csv(prediction_report)
このコードは、予測結果を含むDataFrame (pred) を処理して、予測結果にラベル(「Active」または「Inactive」)を付け、最終的な予測レポートをCSVファイルとして保存する手順を示しています。各ステップは以下の通りです:
- 予測結果DataFrameのインデックス設定:
pred.index = X_vs.index:predDataFrameのインデックスを、予測に使用した入力データ (X_vs) のインデックスと同じに設定します。これにより、予測結果を元のデータと照合しやすくなります。
- 予測結果の結合:
predictions = pd.concat([pred], axis=1):predDataFrameを**predictions**という新しいDataFrameにコピーします。
- 予測値の置換と表示:
for col in ['Prediction', 'AD']: ‘Prediction’列と’AD’列に対してループ処理を行います。predictions[col].replace(0, 'Inactive', inplace=True): 予測値が0のものを「Inactive」に置換します。predictions[col].replace(1, 'Active', inplace=True): 予測値が1のものを「Active」に置換します。print(predictions.head()): 置換後の先頭5行を表示します。
- 予測レポートのCSVファイルへの保存:
predictions.to_csv(prediction_report): 変換された予測結果をCSVファイル(**prediction_report**に指定されたパス)に保存します。
このプロセスにより、数値で表される予測結果がより読みやすい形式(「Active」や「Inactive」のラベル)で表現され、最終的な予測レポートが作成されます。このレポートは、さらなる分析や意思決定プロセスに役立つことが期待されます。
以下のように出力できると思います。
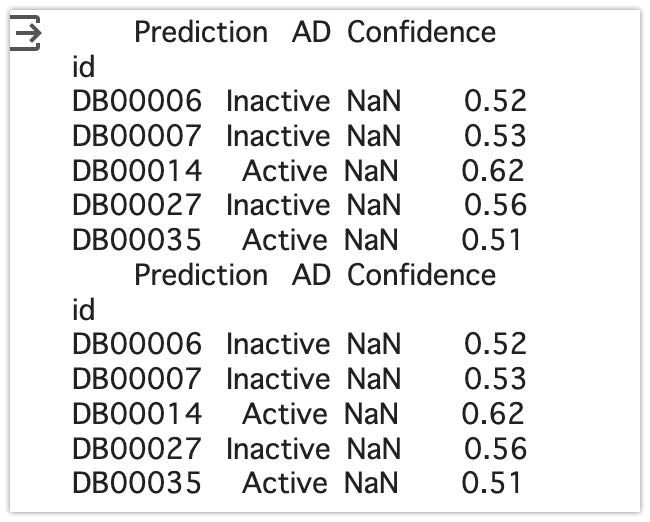
またprediction_report.csvとして、全データを確認できます。
結果
結果を解析してみると、
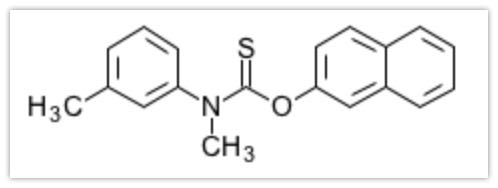
Activeを示したものは220/1520であり、その中で信頼度が0.6以上のものは23個あり、0.7以上のものは0個でした。最も信頼度が高く(0.69)、activeを示したものはDB00525のTolnaftateでした。
最後に
いかがでしたでしょうか?ここまで5章に渡り、データの収集から、整形、モデルの構築、そして、in silicoスクリニーニングを行ってきました。化学構造の扱いや機械学習のあり方など、その基礎を学べたかと思います。今後の方針としては、モデルの改良により精度の高くしたり、違うターゲットにも適用していきたいと思います!また得られた化合物Tolnaftateについて、実際の物性をin silico創薬の様々なツールを使って、情報を集める予定です。
謝辞
この記事は以下の論文のフォローとなります。機械学習とAI創薬についてわかりやすく書かれています。このような論文を書いて下った筆者らに感謝を申し上げます。
NCBI – WWW Error Blocked Diagnostic
この記事を書くにあたり、**@kimisyo(晶 公畑)**さんの記事が大変参考になりました。ありがとうございます。
論文と公共データベースを使って無料で始めるAI創薬 – Qiita
自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1500 今なら50%OFF!!
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1,500 今なら50%OFF!!
分子ドッキングやMDシミュレーションなど、
自宅でできるin silico創薬の解析方法を解説したものになります!

