
分子ドッキングは、薬品や生体分子の相互作用を予測するためのコンピュータシミュレーション手法です。この記事では、分子ドッキングの基本原理や手法、具体的な応用例について解説しています。LZerD pairwise dockingという手法に焦点を当てて紹介しています。
学ぶことによるメリットとしては、新薬開発やタンパク質研究において効果的な薬物スクリーニングや設計が可能となります。また、タンパク質の相互作用解明や薬物の最適化にも役立ちます。さらに、計算科学と生命科学の知識を統合することで、より深い理解と応用が可能となります。
ぜひトライしてみてください!
macOS Ventura(13.2.1)

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1500 今なら50%OFF!!
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1,500 今なら50%OFF!!
分子ドッキングやMDシミュレーションなど、
自宅でできるin silico創薬の解析方法を解説したものになります!
本記事を進むにあたって、PyMOLのダウンロードをお願いします。
大阪大学の蛋白研究所からインストールの仕方が解説されています。
分子ドッキングとは?
分子ドッキングとは、薬品や生体分子(例えばタンパク質)がどのように相互作用するかを予測するコンピュータシミュレーションのことです。
特定のタンパク質と薬物がどのように結合するか、どの部分が互いに触れ合うかを計算し、その結果から薬物の効果を予測します。これは新薬開発などに非常に役立ちます。
現在様々なwebで使うことのできる分子ドッキング手法があります。
今回はその中でも、タンパク質-タンパク質ドッキングを扱うLZerD pairwise dockingについて紹介していきます。
タンパク質の下準備
まず各手法の解析の前にドッキングするタンパク質の下準備をしていきたいと思います。今回はモデルのタンパク質としてProtein Data Bank(PDB)の番号8DHAであるレプチンとその受容体を使います。
レプチンは、脂肪組織に由来するタンパク質ホルモンであり、体重とエネルギーバランスの調節に重要な役割を果たしています。レプチンは脂肪細胞から血液中に放出され、脳の視床下部に存在する特定の受容体に結合することで、食欲を抑制し代謝を調節する作用を持っています。
薬物としてのレプチンの魅力は、肥満治療における可能性です。レプチンの欠乏または抵抗は、肥満の原因となることが知られています。そのため、レプチンの補充や受容体の活性化を促進する薬剤の開発が進められています。
レプチン薬物の魅力は、食欲を抑制する効果と体重減少をもたらす可能性です。これにより、肥満者やメタボリックシンドロームの患者にとって、体重管理や疾患リスクの低減に役立つことが期待されています。さらに、レプチンは食欲を制御する神経回路にも関与しているため、食欲をコントロールする新たな治療法の開発にもつながる可能性があります。
最近ではインスリンを用いずに、レプチンを使って、血糖値を下げることにも成功しており、糖尿病への治療へも期待されています。
(PDB)の番号8DHAであるレプチンとその受容体は結合している箇所のみを実際に取り出したもので、全体は8DH8に登録されています。今回は計算量を少なくするために、番号8DHAであるレプチンとその受容体を使います。
まずPymolのタブのFile→Get PDB…に8DHAと打ち、レプチンとその受容体を表示させます。
続いてDisplay→Sequenceより全体の配列を表示します。
/C/C/1から始まる部位がレプチンに該当するので、この配列を選択します。
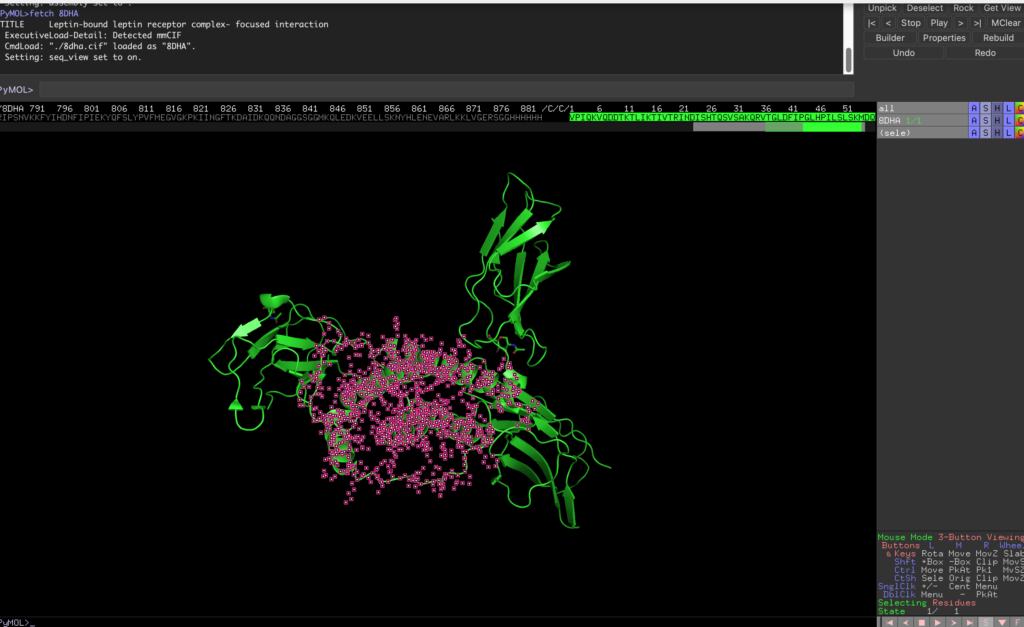
続いて、以下のコードを打ち、実行します。
save leptin.pdb, seleこれによって、選択部位がleptin.pdbとして保存されます。
続いてレプチン以外の表示されているレプチン受容体の部分を選択し、
save focus_leptinR.pdb, seleとして保存します。2-acetamido-2-deoxy-beta-D-glucopyranoseも一緒に結合していますが、こちらの部分は選択しないでください。
これによって、選択部位がfocus_leptinR.pdbとして保存されます。
これらのデータはMacintosh HD/ユーザ/自分のパソコンの名前
の階層に保存されていると思います。
以上でタンパク質の下準備は終了です。
LZerD pairwise dockingとは?
LZerD pairwise dockingは、2つのタンパク質構造(受容体とリガンド)を入力として、それらを組み合わせてタンパク質複合体の3D構造モデルを作成する手法です。まず、可能なすべての相互作用インターフェース領域と相互作用角度をサンプリングして数万のドッキング構造を生成します。次に、生成されたモデルをクラスタリングして絞り込み、最後に3つのスコアリング関数からのスコアランク(ランクサム)の合計によってモデルをランク付けします。これにより、最も可能性の高いタンパク質複合体の3D構造が予測されます。
早速LZerD pairwise dockingのサイトに行ってみましょう。
- Get Startedを選択します。
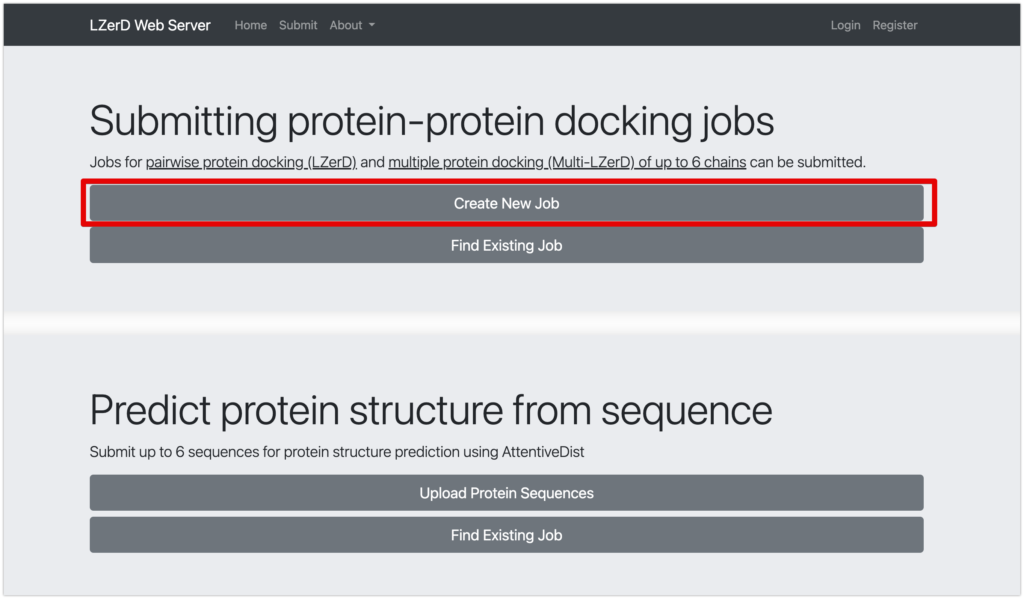
- Submitting protein-protein docking jobsのCreate New Jobを選択します。※1
- Upload Protein 1に
leptin.pdbを選択します。 - Upload Protein 2に
focus_leptinR.pdbを選択します。 - emailを書きます。
- Send Email On Job StartはTrueにチェックを入れます。
- 他の設定はそのままにしておきます。
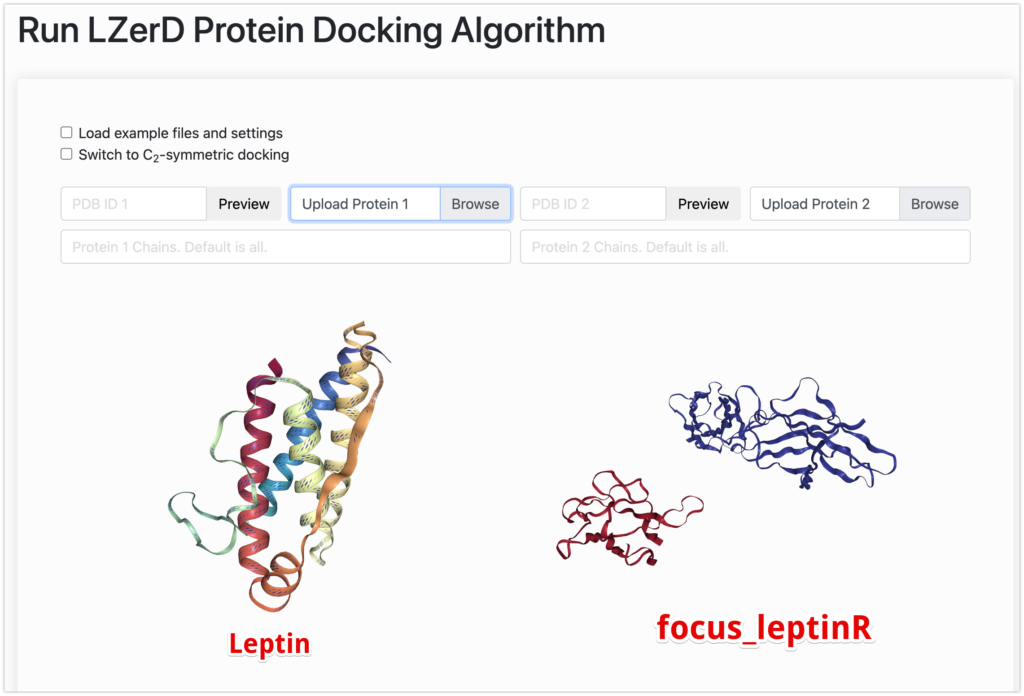
- Submitを押し、しばらくすると、入力したメールに結果が送られてきます。※2
※1
※2
LZerDペアワイズドッキングのメカニズム
LZerD(Local 3D Zernike descriptor-based protein Docking)ペアワイズドッキングは、二つのタンパク質構造(通常は受容体とリガンドと呼ばれます)を入力として取り、それらを組み合わせてタンパク質複合体の3D構造モデルを作成する手法を指します。
このプロセスは主に以下の三つのステップで行われます:
- LZerDは入力として提供された二つの構造を取り、可能なすべての相互作用インターフェース領域と相互作用角度をサンプリングして数万のドッキング構造を作成します。あまりにも多くの原子が衝突する、相互作用領域が小さい、またはインターフェース領域での形状補完性が低いドッキング構造は排除されます。
- 生成されたドッキングモデルは、ユーザー定義のクラスターカットオフ(デフォルトはルート平均二乗偏差、RMSD、4オングストローム)でクラスタリングされます。このステップでは通常、ドッキングモデルが数千から数万まで減少します。
- 残ったモデルは、3つのスコアリング関数(DFIRE、GOAP、ITScore)からのスコアランク(ランクサム)の合計によってランク付けされます。これらのスコアリング関数は、モデル内の原子間相互作用が実験的に決定されたタンパク質構造で観察される距離と角度の特徴に似ているかどうかをチェックします。
このようにして、LZerDペアワイズドッキングは、タンパク質複合体の可能な3D構造を効率的に探索し、予測することができます。
結果
結果は次のようになりました。結果が出るまで1.5日ほどかかりました。
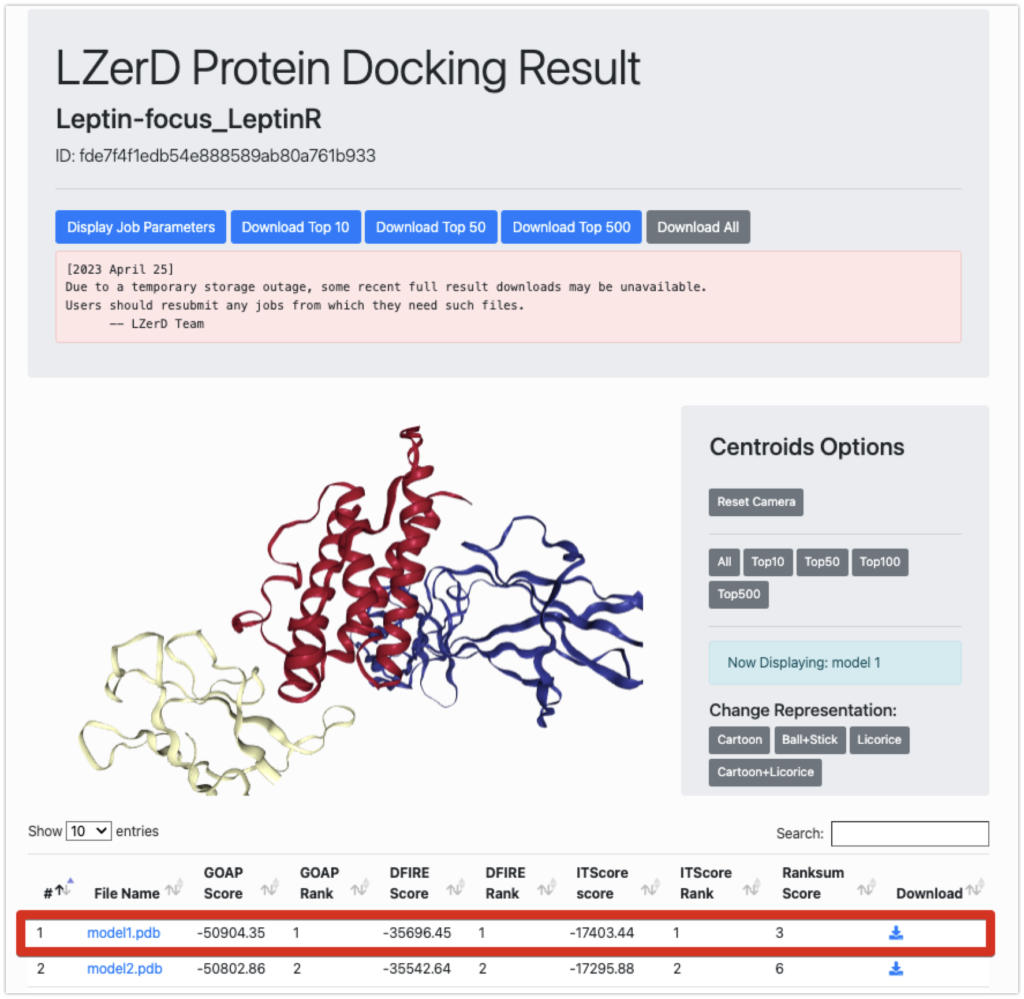
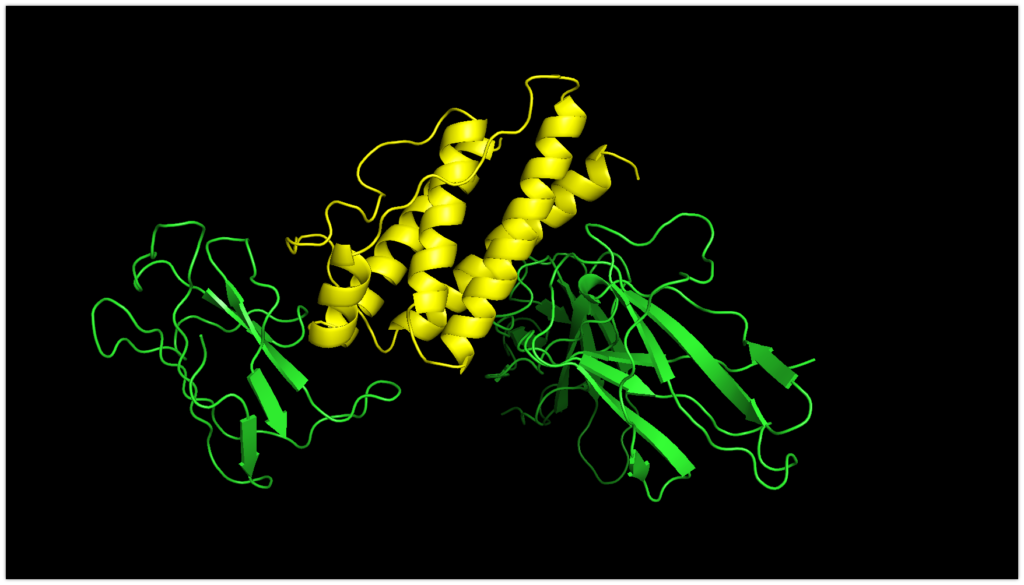
それではランキングが一番低いmodel1.pdbをpymolで見てみましょう。
黄色がLeptin、緑色がfocus_leptinRです。
では元々の構造と比べてみます。
マゼンダで示されているのが元々の構造です。
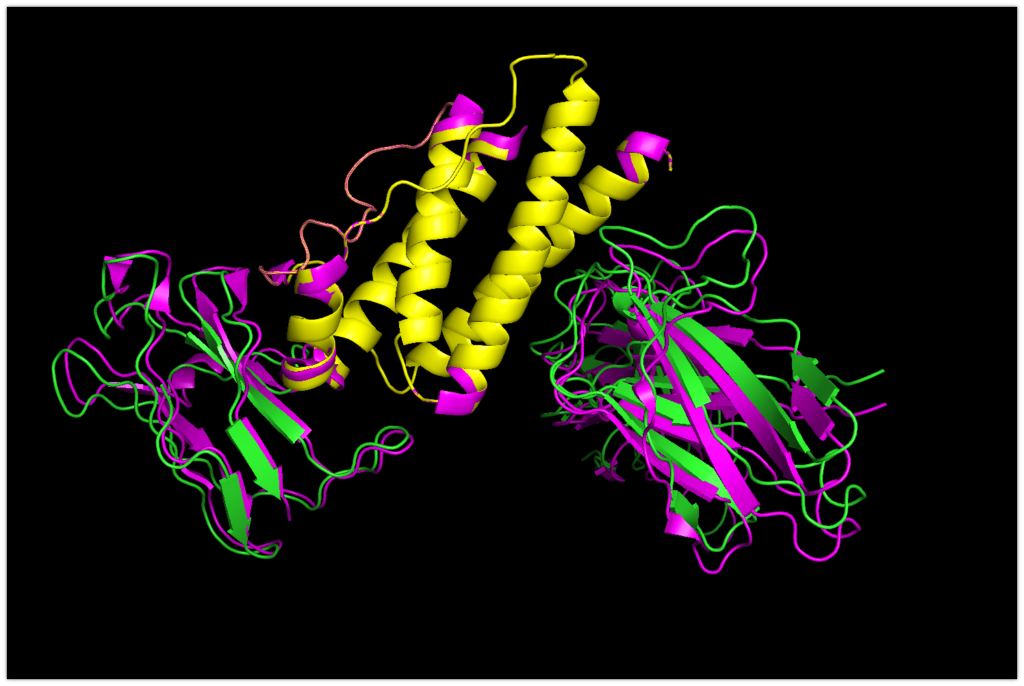
ほぼ決まった位置に結合しているのが分かります。とても精度が良さそうです!
ここでfocus_LeptinRの方が少し位置がずれているのがわかります。
Upload Protein 1にleptin.pdb、Upload Protein 2にfocus_leptinR.pdb
としたため、Upload Protein 1のleptin.pdbは固定したままUpload Protein 2にfocus_leptinR.pdbをドッキングしたからだと思います。
そのため、次はUpload Protein 1にfocus_leptinR.pdb、Upload Protein 2にleptin.pdb としてドッキングさせてみようと思います。
その結果は次のようになりました。
黄緑が今回ドッキングに使ったfocus_leptinR.pdb で、オレンジがドッキングしたleptin.pdb です。
マゼンダで示されているのが元々の配列(PDB:8DHA)です。

focus_leptinR.pdb はずれていないので、やはりUpload Protein 1を固定して、Upload Protein 2をドッキングしてそうですね。それにしても leptin.pdb が精度良くドッキングしていることに驚きです。
最後に
いかがでしたでしょうか?
本記事ではタンパク質ータンパク質ドッキング法の中でLZerD pairwise dockingについて取り上げてみました。
こちらはサーバーで簡単にドッキングを試すことができるので、ぜひ皆さんも試してみてください。
以前の記事ではClusPro、PatchDockを取り上げましたが、こちらとは原理が少し違い、今回のタンパク質では精度良くドッキングできています。
さらにLZerD Web Serverでは
Multi-LZerDの複数チェーンドッキング:3つ以上のタンパク質構造を入力とし、それらを全て組み合わせて複合構造を作成する手法。
AttentiveDistのシングルチェーンタンパク質構造予測:個々のタンパク質配列を入力とし、その構造を新規に予測する手法。
も可能となっています。興味ある方はぜひ試してみてください!


“2023年7月発表された最新のLZerD pairwise dockingという手法”とありますが、2023年7月という日付は参考文献にある書籍(の本手法の紹介記事)が公開された日時であって、手法自体は大分昔に論文が出ているようですしweb serverが公開されたのも2021年ではないでしょうか。手法としては最新のものではなくむしろ古いものだと思うのですが、間違っていたらすみません。
references
https://lzerd.kiharalab.org/about/references/
aaa様
大変貴重なコメントをいただき、誠にありがとうございます。
調査の結果、おっしゃる通り、この手法が開発されたのはもっと前であり、サーバーが構築されたのが2021年で、この手法が記載されて本が出版されたのが2023年7月だということがわかりました。
それにより、記事から”2023年7月発表された最新の”という表現は削除いたしました。
aaa様のおかげで、誤情報が広まる前に記事を修正することができました。心から感謝いたします。
以後このような誤りがないよう、Labcode一同一層の努力してまいります。
引き続きLabcodeをご愛顧いただけるると幸いです。
もし他に何かご連絡いただけることがありましたら、TwitterのDM(https://twitter.com/LabCodeBlog)にてお気軽にやりとりさせていただけたらと思います。どうぞよろしくお願いいたします。