
R言語を使ってバイオインフォマティクスがしたい方は多いのではないでしょうか。実際、RはRNA-seq解析によく使われるプログラミング言語になります。
この記事では、バイオインフォマティクスの中でもRNA-seq解析に必要な最低限のR文法とライブラリの知識について解説します。またRの特徴であるデータフレームの扱い方についてサンプルコードを交えて初心者向けに解説します。
本記事を読んで、RNA-seq解析におけるR言語を始める第一歩を踏み出してみましょう。
macOS Monterey(12.4), クアッドコアIntel Core i7, メモリ32GB

公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます
R言語とは?
R言語は、統計分析やグラフ作成などのために開発されたプログラミング言語です。統計学や生物学向けに大量のパッケージが存在しております。R言語は文法が簡潔であり、グラフ作成の機能が強力なことが特徴です。また、他の言語とのインターフェイスも提供されており、大規模なデータ処理やモデリングを行うこともできます。
またRはRNA-seq解析においても利用されます。RNA-seq解析パッケージを利用すると、遺伝子発現量の比較、差分発現解析、GO領域やKEGGパスウェイ分析などが簡単に行えるようになっています。また、可視化機能も豊富で、発現量のヒストグラムや発現量の比較などをグラフとして出力することもできます。
Rstudioの設定
Rを実行するにははじめに環境構築が必要となります。今回はRを実行する環境としてRstudioを使っていきます。
各OS環境を元にこちらのサイトよりインストーラーをダウンロードしてください。https://cran.r-project.org/
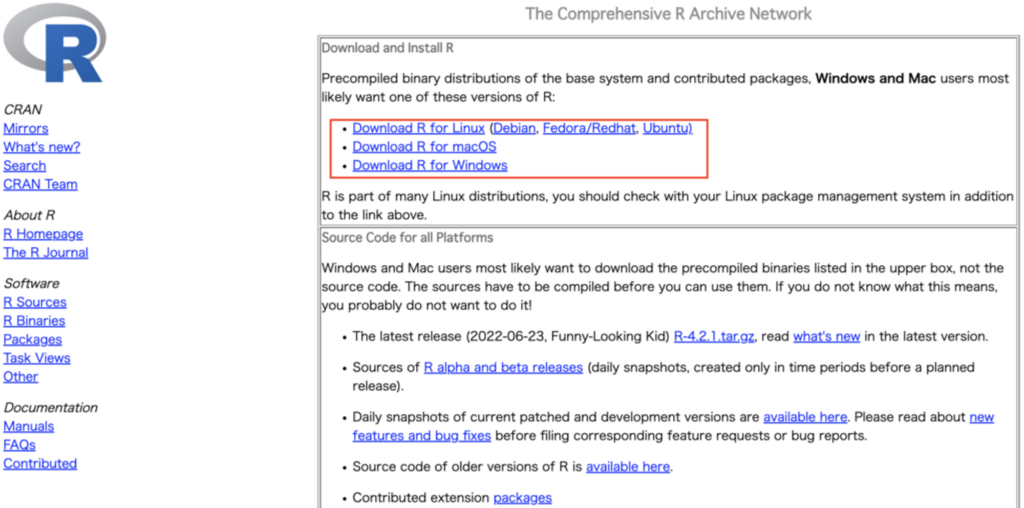
インストールするバージョンは最新版を選んでください。一番トップにあるものが最新版です。

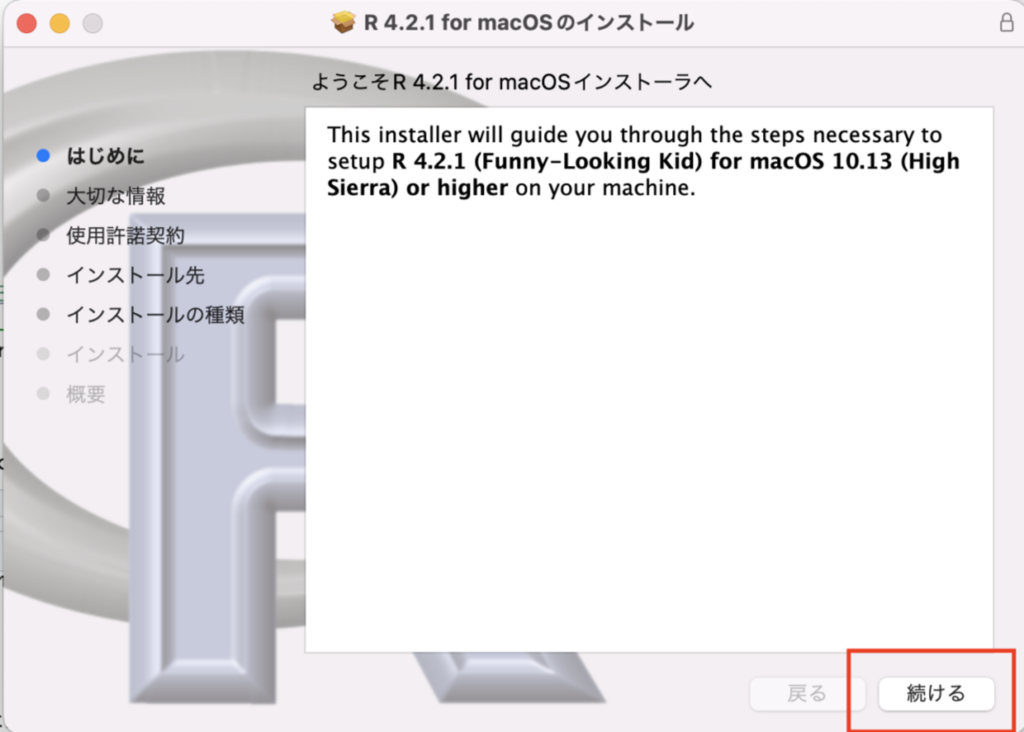
インストール画面では「続ける」を押して完了させてください。
Rstudioでは左上のエリアにコードを書いていきます。Runを押すと行実行ができます。
Sourceを押すとスクリプト全体が実行できますので覚えておいてください。
RパッケージマネージャBioconductorについて
生物化学のデータ解析のためのR言語パッケージのコレクションです。
約数百種類のパッケージが管理されており、Bioconductorを通じてインストールすることができます。
バイオ系でR言語がよく使われる理由はなんといってもそのライブラリの充実度だと思います。しっかり抑えていきましょう。
パッケージの呼び出しは library 関数で実施できます。
library(clusterProfiler)しかし、大抵のライブラリはインストールが必要です。R言語でパッケージをインストールするには、CRANのinstall.packages()関数を使用します。
install.packages() Bioconductorだけで公開されているものはBiocManager パッケージの install 関数を使ってインストールする必要があり、BiocManager::install()関数にてインストールができます
install.packages("BiocManager") # はじめにBiocManagerをインストールする
library(BiocManager) # インストールしたBiocManagerを使える状態にする
install("edgeR") # edgeRをBioconductorからインストールするR言語の基本的な文法
では実際に、RNA−seq解析に必要な最低限のRコードを使ってみましょう。
出力結果をコメントアウトで書くので、ペーストする時はコメントアウトを含めないように気をつけてください。
まずは、入力をそのまま出力してみましょう。
print("hello")
#出力結果
#[1] "hello"“hello”と出力できましたね!””がないと、きちんと出力できないので気をつけてください。
変数宣言するには<-を使います。
x <- 5
x
#出力結果
#[1] 5R言語の最も重要な特徴はベクトル定義 です。 ベクトルは連なったデータの入れ物です。
ベクトルを作成するにはc()関数で値を結合します。
y <- c("apple", 1, "orange", 2, "banana")
y
#出力結果
#[1] "apple" "1" "orange" "2" "banana"for( X in Y ) { ~~~~ }でループを回すことができます。 c()で作ったベクトルを一つずつ出すときにも利用するので覚えておいてください。
for(i in 1:10){
print(i)
}
#出力結果
#[1] 1
#[1] 2
#[1] 3
#[1] 4
#[1] 5
#[1] 6
#[1] 7
#[1] 8
#[1] 9
#[1] 10if( ){ ~~~ } else { ~~~ }で条件分岐を作成できます。
num <- 4
if(num %% 2 == 0) {
print(paste(num, "は偶数です"))
} else {
print(paste(num, "は奇数です"))
}
#出力結果
# [1] "4 は偶数です"typeof() で型の確認ができます。Rでは中に入っている型の確認をしますので覚えておいてください。
ちなみにclass()関数でも似たようなことができます。
y <- "apple"
print(typeof(y))
#出力結果
#[1] "character"R言語でデータフレーム操作
Rの特徴としてデータフレーム操作ができることが挙げられます。データフレームとして扱うためにRのようなデータ分析言語を利用しますのでしっかり覚えていきましょう。R言語でデータフレーム(表形式のデータ)を操作するには、以下のように書きます。
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"))
print(df)
# 出力結果
col1 col2
1 1 A
2 2 B
3 3 Chead()関数を使用するとデータフレームの中身をデフォルトで6行表示させることができます。データフレームの中身を見たいときに多用します。引数で表示する行数を指定するとその分の行数が出力されます。
# データフレームを作成
df <- data.frame(col1 = c(1, 2, 3, 4, 5, 6, 7, 8, 9, 10),
col2 = c("A", "B", "C", "D", "E", "F", "G", "H", "I", "J"),
col3 = c(TRUE, FALSE, TRUE, TRUE, FALSE, FALSE, TRUE, TRUE, TRUE, FALSE))
# デフォルトで6行表示
head(df)
# 出力結果
col1 col2 col3
1 1 A TRUE
2 2 B FALSE
3 3 C TRUE
4 4 D TRUE
5 5 E FALSE
6 6 F FALSE
--------------------------------------------
# 引数で表示する行数を指定して表示
head(df, 3)
# 出力結果
col1 col2 col3
1 1 A TRUE
2 2 B FALSE
3 3 C TRUE行・列の操作として df[row, col]またはdf$colを使用して、特定の行・列を選択することができます。
# データフレームを作成
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"), col3 = c(TRUE, FALSE, TRUE))
# 1行目を選択
row1 <- df[1, ]
# col2列を選択
col2 <- df$col2
#出力結果1
> row1
col1 col2 col3
1 1 A TRUE
#出力結果2
> col2
[1] "A" "B" "C"行・列の削除: df[-row, -col]を使用して、特定の行・列を削除します。指定した行・列に-をつけると削除できます。
# データフレームを作成
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"), col3 = c(TRUE, FALSE, TRUE))
#出力結果
col1 col2 col3
1 1 A TRUE
2 2 B FALSE
3 3 C TRUE
-------------------------------------------------
# 1行目と3列目を削除
df <- df[-1, -3]
#出力結果
col1 col2
2 2 B
3 3 Cデータフレームの出力結果を見比べると、1行目と3列目が削除されているかと思います。
データフレームの特定の列だけを取り出したいときがあります。そういうときは、df[, c("col2")] のようにc(”[列名]”)で指定してあげます。
# データフレーム作成
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"), col3 = c(TRUE, FALSE, TRUE))
# col2だけを取り出す
df <- df[, c("col2")]
# 出力結果
col2
1 A
2 B
3 Ccolnames(df)を使うと、カラム名が取得できます。
# データフレームを作成
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"), col3 = c(TRUE, FALSE, TRUE))
# 列名を出力
colnames(df)
# 出力結果
[1] "col1" "col2" "col3"RNA−seq解析をしているとカラム名をサンプル名にしたい場合が結構あります。 colnames(df) <- c("col1", "col2", ...) でカラム名が変更できるので覚えておいてください
# データフレームを作成
df <- data.frame(col1 = c(1, 2, 3), col2 = c("A", "B", "C"), col3 = c(TRUE, FALSE, TRUE))
# 列名を変更
colnames(df) <- c("SampleA", "SampleB", "SampleC")
# 出力
print(colnames(df))
# 出力結果
[1] "SampleA" "SampleB" "SampleC"ここまでデータフレームの扱い方について様々触れてきましたが、RNA-seq解析をする上で外部のファイル(csv, tsv, excel)を読み出して RNA-seq解析するパターンが多いです。試しにcsvファイルを読み出してみましょう。
以下のファイルをdesktopにダウンロードしてきてください。
ちなみにsample.csvの中身はこの様になっています。
id,name,age
1,Alice,25
2,Bob,30
3,Charlie,35
4,David,40
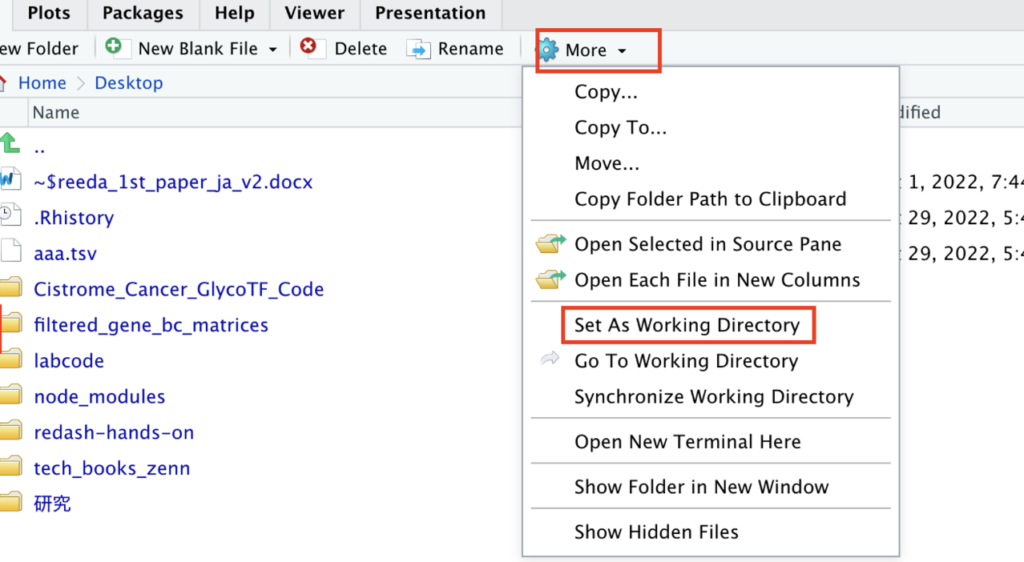
5,Eve,45デスクトップにsample.csvをダウンロードしてきたら、Rstudioの右下画面で
- Desktopを選ぶ
- Moreをクリック
- Set As Working Directory
を順に選びます。この作業を間違うと後のコードが一切通らなくなるのでお気をつけください。
では、実際にcsvファイルを読み出してみましょう。csvファイルを読み出すときはreadrというライブラリを指定する必要があります。
# readrライブラリの読み出し
library(readr)
# CSVファイルの読み込み
df <- read_csv("sample.csv")
#出力結果
# A tibble: 5 × 3
id name age
<dbl> <chr> <dbl>
1 1 Alice 25
2 2 Bob 30
3 3 Charlie 35
4 4 David 40
5 5 Eve 45データフレームをcsvファイルから読み出せました。read_csv関数を用いてcsvファイルを読み出すと、tidyverse というパッケージに含まれるデータ構造であるtibble で読み出されます。
これはこれで扱えるのですが、先程のようなデータフレームで扱いたかったら、as.data.frame を併用すると良いです。
df <- as.data.frame(read_csv("sample.csv"))
df
#出力結果
id name age
1 1 Alice 25
2 2 Bob 30
3 3 Charlie 35
4 4 David 40
5 5 Eve 45これでcsvファイルの読み出しはできるようになりました。tsvファイルを読み出すときはread_csvの代わりにread_tsv を使ってください。
# readrライブラリの読み出し
library(readr)
# CSVファイルの読み込み
df <- read_csv("sample.tsv")エクセルファイルを読み出すときはreadrライブラリの代わりにreadxl ライブラリを使ってください。
# readxlライブラリの読み出し
library(readxl)
# エクセルファイルの読み込み
df <- read_excel("example.xlsx")最後に
いかがだったでしょうか。思ったよりも簡単にR言語が使えるかと思います。次回はggplotを用いた作画を扱いと思いますのでぜひともそちらもご閲覧ください。
公共データを用いたRNA-seq解析に関する初心者向け技術書を販売中
¥2,500 → ¥1,500 今なら40%OFF!!
画面キャプチャをふんだんに掲載したわかりやすい解説!
自宅PCからドライ研究が始められます

