

scRNA-seqデータで擬似バルク解析(pseudo bulk analysis)を行う方法をご存知でしょうか。擬似バルク解析を使うと、シングルセルRNA-seqデータを用いて、bulk RNA-seqの解析手法を適用できるようになります。とても便利な手法なのでぜひチャレンジしてみましょう。
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB


公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!

公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!


TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます
擬似バルク RNA-seq 解析(pseudo bulk RNA-seq analysis)とは?
疑似バルク解析(pseudo bulk analysis)とは、シングルセルRNA-seqデータを用いて、bulk RNA-seqの解析手法を適用できるようにすることです。以下でもう少し詳しく説明します。
バルクRNA-seqとは
バルクRNA-seqは、多数の細胞から抽出されたRNAをまとめて解析します。比較的安価で、広く利用されています。scRNA-seqと違って全体のRNA発現プロファイルを提供しますが、個々の細胞の違いは反映されません。そのため大規模な発現変動の解析、遺伝子発現のパターンの特定、疾患関連遺伝子の発見などに適しています。
疑似バルク解析とは
疑似バルク解析(pseudo-bulk analysis)は、シングルセルRNA-seq(scRNA-seq)データを用いて、バルクRNA-seq解析に似た方法でデータを解析する手法です。具体的には、個々の細胞から得られた発現データを、特定のグループ(例えば同じ細胞タイプや状態)ごとに合計または平均して解析を行います。これにより、シングルセルレベルの詳細な情報を保持しつつ、バルクRNA-seq解析の利点を活かすことができます。イメージとしては以下の図が分かりやすいと思います。
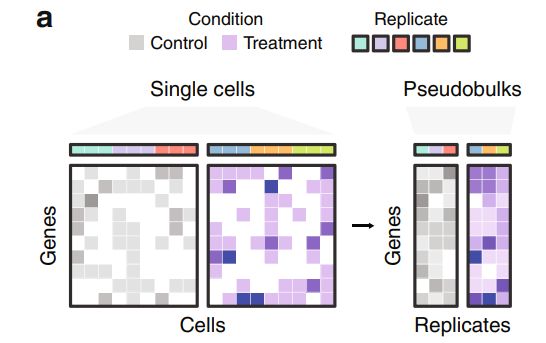
Jordan W. Squair et al., 2021 https://www.nature.com/articles/s41467-021-25960-2
擬似バルク RNA-seq 解析(pseudo bulk RNA-seq analysis)の実装方法
以上述べたことを基に、疑似バルク解析をRでコーディングしてみます。以下に実装例を示します。
library(ExperimentHub)
library(Seurat)
library(DESeq2)
library(tidyverse)
# get data
eh <- ExperimentHub()
query(eh, "Kang")
sce <- eh[["EH2259"]]
seu.obj <- as.Seurat(sce, data = NULL)
# get mito percent
seu.obj$mitoPercent <- PercentageFeatureSet(seu.obj, pattern = '^MT-')
# filter
seu.filtered <- subset(seu.obj, subset = nFeature_originalexp > 200 & nFeature_originalexp < 2500 & nCount_originalexp > 800 & mitoPercent < 5 & multiplets == 'singlet')
# run Seurat's standard workflow steps
seu.filtered <- NormalizeData(seu.filtered)
seu.filtered <- FindVariableFeatures(seu.filtered)
seu.filtered <- ScaleData(seu.filtered)
seu.filtered <- RunPCA(seu.filtered)
ElbowPlot(seu.filtered)
seu.filtered <- RunUMAP(seu.filtered, dims = 1:20)
# visualize
cell_plot <- DimPlot(seu.filtered, reduction = 'umap', group.by = 'cell', label = TRUE)
cond_plot <- DimPlot(seu.filtered, reduction = 'umap', group.by = 'stim')
cell_plot|cond_plot
# pseudo-bulk workflow
seu.filtered$samples <- paste0(seu.filtered$stim, seu.filtered$ind)
DefaultAssay(seu.filtered)
cts <- AggregateExpression(seu.filtered, group.by = c("cell", "samples"), assays = 'originalexp', slot = "counts", return.seurat = FALSE)
cts <- cts$originalexp
# transpose
cts.t <- t(cts)
# convert to data.frame
cts.t <- as.data.frame(cts.t)
# get values where to split
splitRows <- gsub('_.*', '', rownames(cts.t))
# split data.frame
cts.split <- split.data.frame(cts.t, f = factor(splitRows))
# fix colnames and transpose
cts.split.modified <- lapply(cts.split, function(x){ rownames(x) <- gsub('.*_(.*)', '\\1', rownames(x)) t(x)
})
# 1. Get counts matrix
counts_bcell <- cts.split.modified$`B cells`
# 2. generate sample level metadata
colData <- data.frame(samples = colnames(counts_bcell))
colData <- colData %>% mutate(condition = ifelse(grepl('stim', samples), 'Stimulated', 'Control')) %>% column_to_rownames(var = 'samples')
# perform DESeq2 --------
# Create DESeq2 object
dds <- DESeqDataSetFromMatrix(countData = counts_bcell, colData = colData, design = ~ condition)
# filter
keep <- rowSums(counts(dds)) >=10
dds <- dds[keep,]
# run DESeq2
dds <- DESeq(dds)
# Check the coefficients for the comparison
resultsNames(dds)
# Generate results object
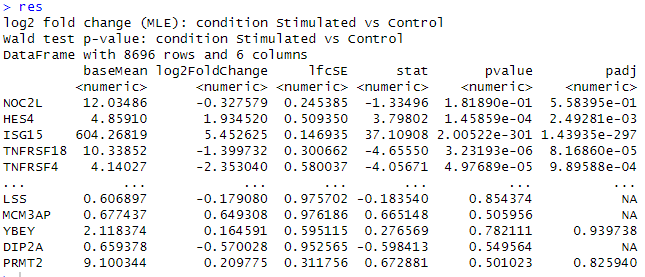
res <- results(dds, name = "condition_Stimulated_vs_Control")
res実行結果
最終的に以下のような出力が出れば成功です!
コードの解説
library(ExperimentHub)
library(Seurat)
library(DESeq2)
library(tidyverse)まず必要なライブラリを読み込みます。ExperimentHubは、バイオインフォマティクス分野で使用される大規模なデータセットを簡便に取得・利用するためのRパッケージです。Bioconductorプロジェクトの一部として提供されており、主にゲノム解析やトランスクリプトーム解析、エピゲノム解析などに関連するデータセットを取り扱っています。
eh <- ExperimentHub()“ExperimentHub”関数を使用してデータを取得します。
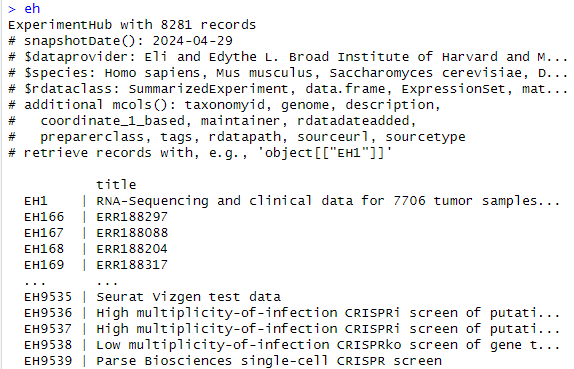
ehの中身を見てみると上記のような出力が確認できます。最初から解説していきます。
ExperimentHub with 8281 records :”ExperimentHub” オブジェクトには 8281 件のレコードが含まれていることが示されています。
snapshotDate(): 2024-04-29:データのスナップショット日付を示しています。2024年4月29日時点のデータです。
dataprovider:データ提供者の情報を示しています。ここでは Eli and Edythe L. Broad Institute of Harvard and MIT などの複数の機関が含まれています。
species:データに含まれる生物種を示しています。
dataclass:データのクラス(データ型)を示しています。
additional mcols():追加のメタデータ列を示しています。
EH1: “RNA-Sequencing and clinical data for 7706 tumor samples from The…”
上記のEH1はExperimentHub中のIDを示しています。そしてtitleはそのデータのタイトルやIDを示しています。
query(eh, "Kang")“query”関数を使用してデータを検索します。上記コードはExperimentHubデータから”Kang”によるデータを検索します。このコードを実行すると以下のような出力が得られます。
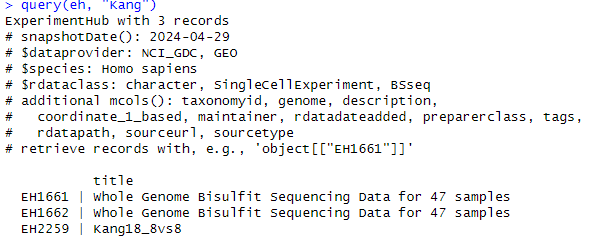
出力を見るとExperimentHubライブラリには”Kang”によるデータが以下の3種類含まれていることが分かります。
データは ID:title の形式で示されています。今回はIDが”EH2259”、タイトルが”Kang18_8vs8”というデータを使用します。
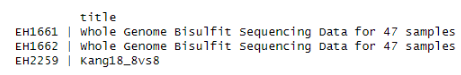
sce <- eh[["EH2259"]]object[[ID名]]とするとデータを取得することができます。今回は使用するデータのIDが”EH2259”なので引数としてそのIDを設定します。データの形式は”SingleCellExperiment”クラスです。
seu.obj <- as.Seurat(sce, data = NULL)”SingleCellExperiment”クラスから”Seurat”クラスに変換します。
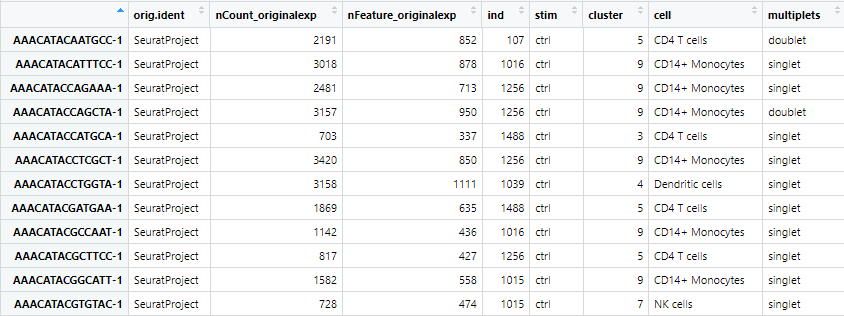
ここまでのデータを View(seu.obj@meta.data) で確認してみると以下のような形式になっています。
seu.obj$mitoPercent <- PercentageFeatureSet(seu.obj, pattern = '^MT-')
seu.filtered <- subset(seu.obj, subset = nFeature_originalexp > 200 & nFeature_originalexp < 2500 & nCount_originalexp > 800 & mitoPercent < 5 & multiplets == 'singlet')“PercentageFeatureSet”関数を使用してミトコンドリア遺伝子の割合を計算します。
この関数は、遺伝子名が「MT-」で始まるミトコンドリア遺伝子を特定し、その発現割合を計算します。計算された割合は新しいメタデータ列としてSeuratオブジェクトに追加され、”seu.obj$mitoPercent”に保存されます。
次に、”seu.obj”オブジェクトから特定の基準に基づいてデータを抽出し、新しいSeuratオブジェクト”seu.filtered”に保存します。
このフィルタリング基準では、検出された遺伝子の数が200から2500の範囲内にある細胞を選択し、発現カウント数が800以上の細胞を選びます。そしてミトコンドリア遺伝子の発現割合が5%未満の細胞を選択します。最後に、”multiplets”列が”singlet”である細胞、すなわちシングルセルとして識別された細胞のみを選びます。これにより、二重細胞(複数の細胞が一つのドロップレットに入っている状態)を除外します。
seu.filtered <- NormalizeData(seu.filtered)
seu.filtered <- FindVariableFeatures(seu.filtered)
seu.filtered <- ScaleData(seu.filtered)
seu.filtered <- RunPCA(seu.filtered)
ElbowPlot(seu.filtered)
seu.filtered <- RunUMAP(seu.filtered, dims = 1:20)このコードでは、シングルセルRNAシーケンシングデータの前処理および可視化のプロセスを示しています。
まず、Seuratオブジェクトの”seu.filtered”に対して正規化を行います。”NormalizeData”関数を使用して、各細胞内の遺伝子発現を正規化し、技術的なばらつきを補正します。
次に、”FindVariableFeatures”関数を使って変動遺伝子を特定し、解析に特に重要な遺伝子を選別します。
次に”ScaleData”関数を用いて、各遺伝子の発現量を平均0、標準偏差1に変換し、異なる遺伝子の発現量を比較しやすくします。
その後、主成分分析(PCA)を”RunPCA”関数で実行し、高次元の遺伝子発現データを低次元の主成分空間に変換します。これにより、データの主要な変動パターンを捉えることができます。
主成分分析の結果を評価するために、”ElbowPlot”関数を使用してエルボープロットを作成します。エルボープロットでは、各主成分の寄与度を視覚化し、解析に適した主成分数を決定するのに役立ちます。
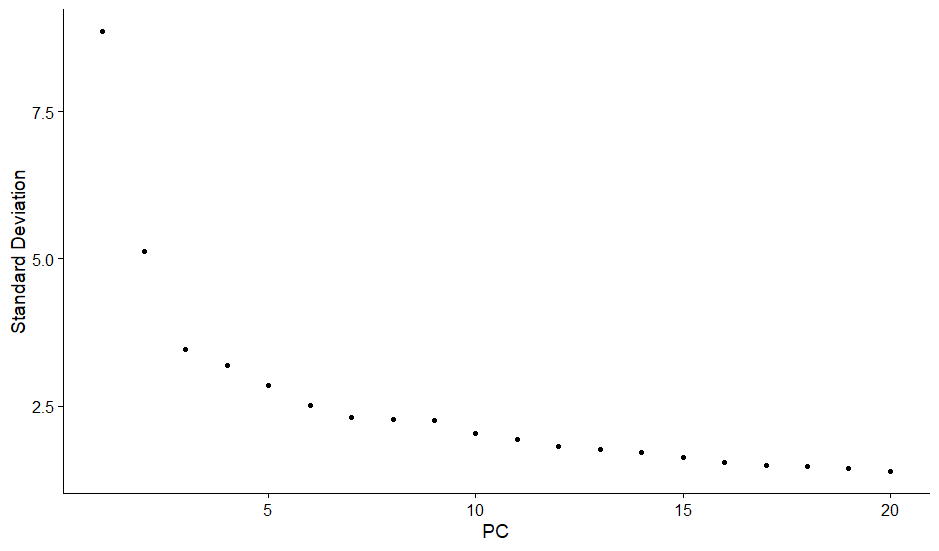
次に、PCAの結果を基に、UMAP(Uniform Manifold Approximation and Projection)を”RunUMAP”関数で実行し、データをさらに低次元空間に埋め込みます。これにより、データのクラスタリングパターンをより明確に視覚化できます。
# visualize
cell_plot <- DimPlot(seu.filtered, reduction = 'umap', group.by = 'cell', label = TRUE)
cond_plot <- DimPlot(seu.filtered, reduction = 'umap', group.by = 'stim')
cell_plot
cond_plot最後に、UMAPの結果をプロットします。”DimPlot”関数を使って、細胞のクラスタリングを視覚化します。結果として得られたグラフを以下に示します。
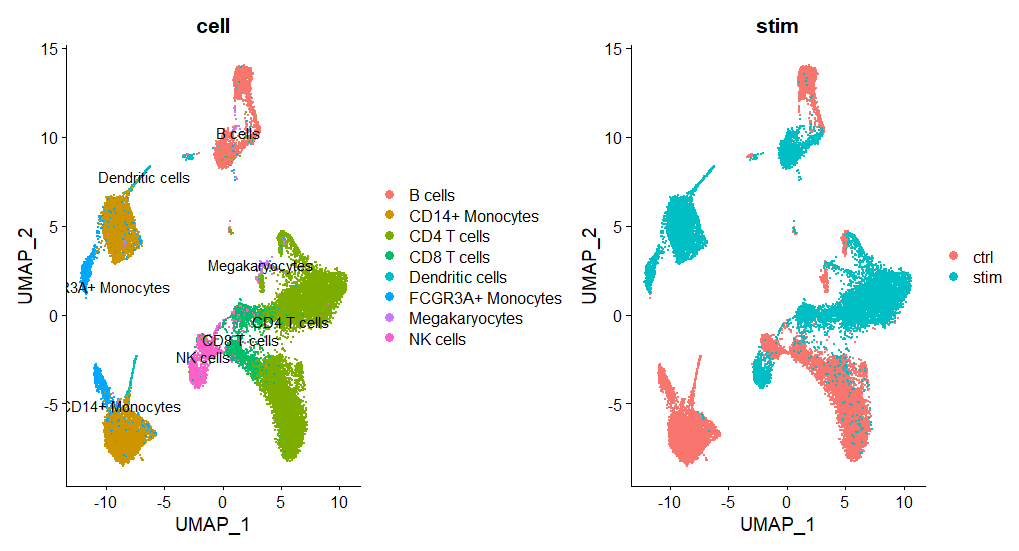
seu.filtered$samples <- paste0(seu.filtered$stim, seu.filtered$ind)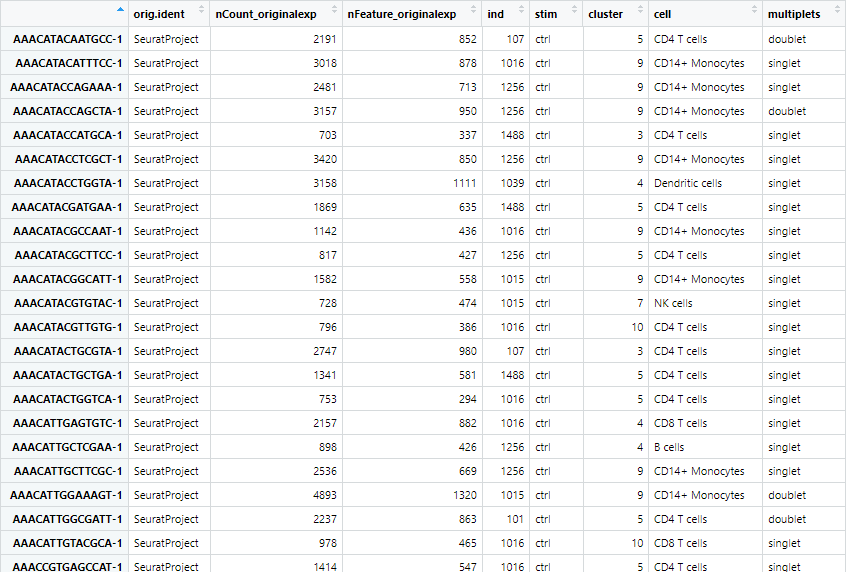
上記の表中の”ind”,”stim”を使用し”sample”という新たな列を作成します。コードを実行すると以下のようになります。
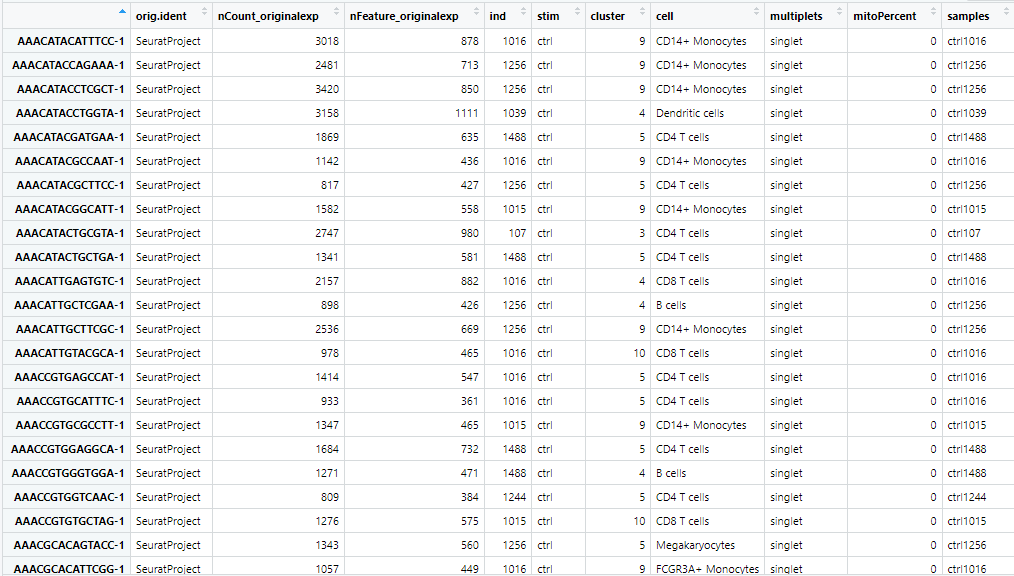
DefaultAssay(seu.filtered)
cts <- AggregateExpression(seu.filtered, group.by = c("cell", "samples"), assays = 'originalexp', slot = "counts", return.seurat = FALSE)
cts <- cts$originalexp“DefaultAssay”関数はオブジェクトの実験データタイプを返す関数です。”AggregateExpression”関数は”group.by”オプションで指定された列をもとに、データを集計します。
上記コードを実行すると”cts”は以下のような構造になります。
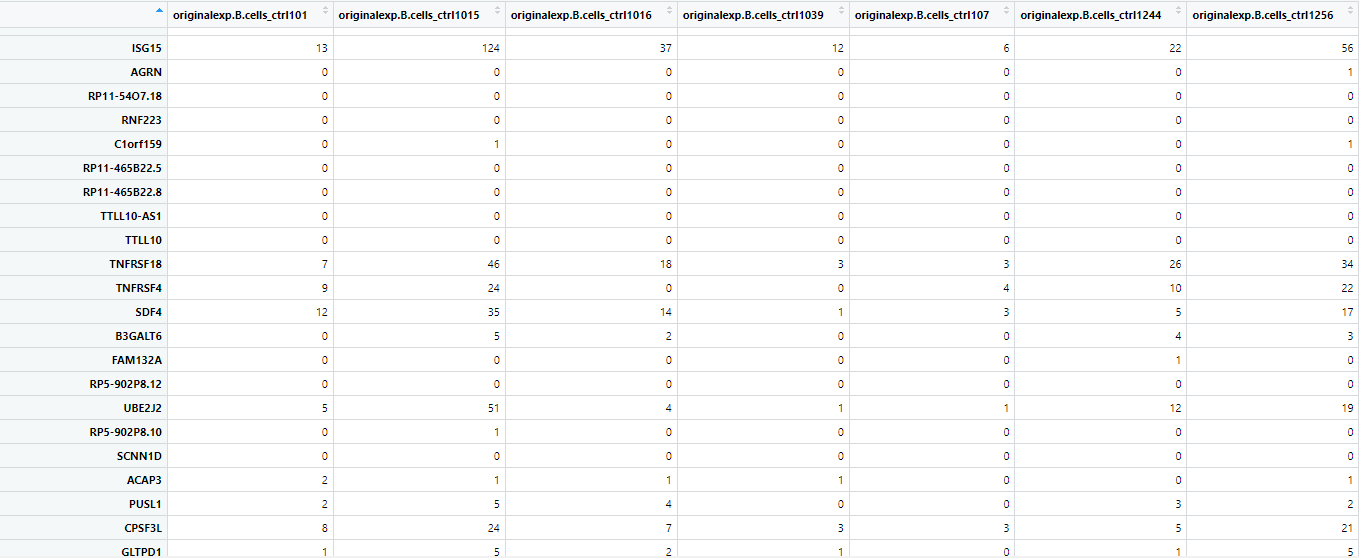
行は遺伝子を、列はサンプルを表しています。
cts.t <- t(cts)
cts.t <- as.data.frame(cts.t)使いやすい形にするため転置した後データフレームにします。
splitRows <- gsub('_.*', '', rownames(cts.t))
cts.split <- split.data.frame(cts.t, f = factor(splitRows))“gsub”関数で行名から細胞タイプを抽出した後、それを基に細胞タイプごとのデータフレームを作成します。
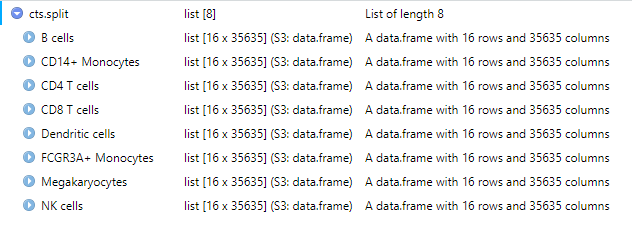
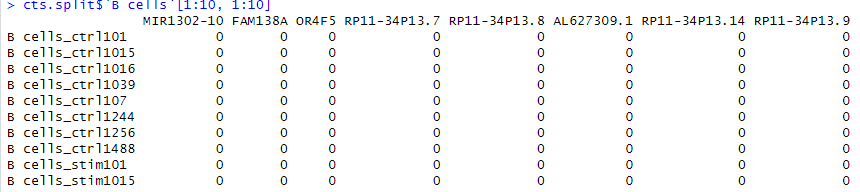
cts.split.modified <- lapply(cts.split, function(x){ rownames(x) <- gsub('.*_(.*)', '\\1', rownames(x)) t(x)
})このコードは、リストに格納されたデータフレームの行名を修正し、データを転置するための処理を行っています。
具体的には、cts.splitというリスト内の各データフレームに対して、行名の変更と転置を適用しています。この処理の詳細を順を追って説明します。
まず、lapply関数を使ってcts.splitリスト内の各データフレームに対して同じ処理を適用します。lapplyは、リストの各要素に対して指定した関数を適用し、その結果を新しいリストとして返します。この場合、匿名関数(function(x) {…})を使用して、各データフレームxに対して以下の操作を行います。
最初に、rownames(x) <- gsub(‘.*_(.*)’, ‘\\1’, rownames(x))という行で、各データフレームの行名を修正します。
具体的には、gsub関数を使って行名に含まれる特定のパターンを置換します。正規表現’.*_(.*)’は、行名の中で最後のアンダースコア以降の文字列(.*_の部分を削除し、.*に一致する部分を取り出す)にマッチします。
置換パターン’\\1’は、マッチした部分のグループ(括弧内の文字列)を指します。これにより、行名の最後のアンダースコア以降の部分のみが新しい行名として設定されます。
次に、t(x)という行で、データフレームxを転置します。転置とは、行と列を入れ替える操作です。これにより、元のデータフレームの行が列になり、列が行になります。
この処理をリスト内のすべてのデータフレームに適用した結果、新しいリストcts.split.modifiedが得られます。
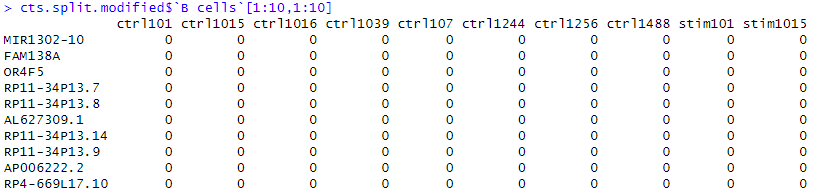
counts_bcell <- cts.split.modified$`B cells`cts.split.modifiedの中から「B cells」という名前の要素を抽出し、そのデータを”counts_bcell”という新しい変数に代入します。”counts_bcell”は以下のような構造になっています。
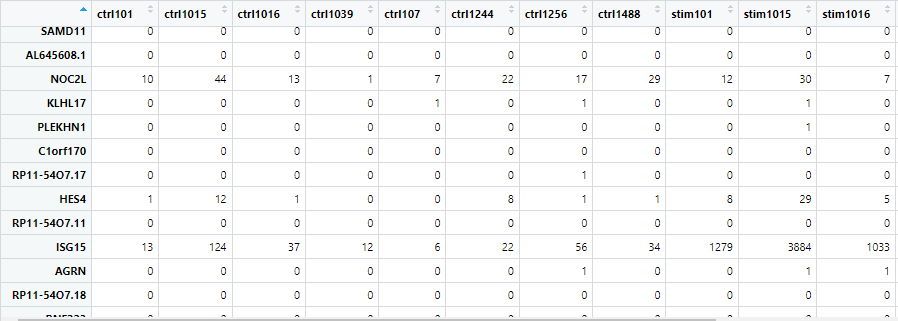
colData <- data.frame(samples = colnames(counts_bcell))先ほど作成した“counts_bcell”の列名(つまり、各サンプルの名前)を抽出して、colDataという名前の新しいデータフレームを作成します。以下のような出力を得られます。
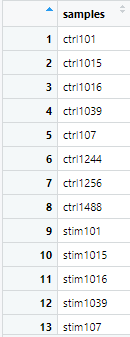
colData <- colData %>% mutate(condition = ifelse(grepl('stim', samples), 'Stimulated', 'Control')) %>% column_to_rownames(var = 'samples')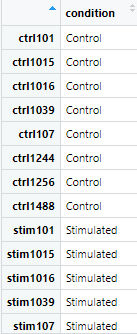
mutate関数によって”sample”列の値が”stim”である場合は”stimlated”、もしそうでないならば”control”として”condition”列を作成します。その後”column_to_rownames”関数によって”sample”列の値を行名にします。
dds <- DESeqDataSetFromMatrix(countData = counts_bcell, colData = colData, design = ~ condition)DEG解析の準備(DESeqDataSetオブジェクトの作成)を行います。
少し前に作成したサンプルごとに遺伝子の発現状況を示す”counts_bcell”、サンプルの状態を示す”colData”を用いて行います。
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]カウントデータが10以下の遺伝子をフィルタリングします。
dds <- DESeq(dds)
resultsNames(dds)DEseqを行い”dds”に結果を代入します。その後”resultsNames”関数によって参照可能な結果の一覧を表示します。
res <- results(dds, name = "condition_Stimulated_vs_Control")
res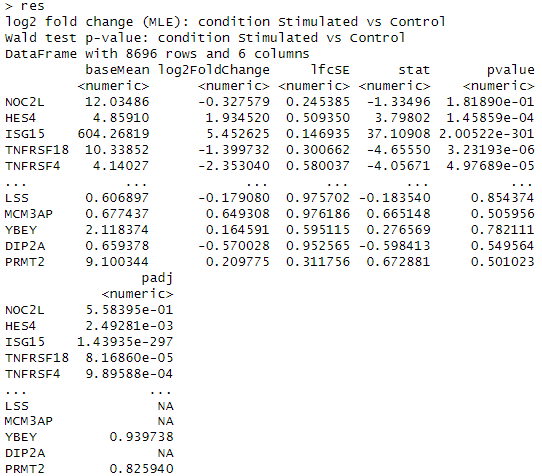
results関数を用いて結果を表示します。上記のような結果が出力されれば成功です。
各列の解説をします。
baseMean: 各条件(ここではStimulatedとControl)での遺伝子の平均発現量です。発現量は通常、条件間での差次的発現解析の基準として使用されます。
log2FoldChange: ログ2スケールでのフォールド変化を示します。これは、Stimulated条件とControl条件の間で遺伝子発現量がどの程度変化したかを示します。正の値は発現量の増加、負の値は減少を意味します。
lfcSE: ログ2フォールド変化の標準誤差です。これは、log2FoldChangeの推定値の不確実性を示します。
stat: Wald検定の統計量です。これは、各遺伝子のlog2FoldChangeがゼロであるという帰無仮説をテストするための値です。
pvalue: Wald検定に基づくp値です。これは、帰無仮説(log2FoldChangeがゼロである)を棄却するかどうかを判断するために使用されます。小さいp値は、条件間で有意な発現変化があることを示します。
padj: p値を複数検定の影響を考慮して調整したものです。調整後のp値は、False Discovery Rate(FDR)を制御するために使用されます。一般に、0.05未満のpadj値は統計的に有意とみなされます。
最後に
いかがだったでしょうか。scRNA-seqデータを用いた疑似バルク解析ができるとscRNA-seqデータ解析の幅が広がると思います。ぜひ挑戦してみてください。
公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!
TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます
参考
scRNAseqとbulk RNAseqの違いを説明した時に用いた図の元論文

