

この記事では、自分のパソコンで行えるMDシミュレーションに焦点を当てており、具体的にはGROMACSというソフトウェアを使用してタンパク質-低分子リガンドの動きをシミュレーションします。この記事を学ぶことで、タンパク質と低分子リガンドの動力学的な挙動や相互作用を理解することができます。ぜひ、トライしてみてください!
本記事のGROMACSのタンパク質-低分子リガンドシミュレーションは設定が長いので、前編と後編に分かれています。後編はこちらから。
本記事を進むにあたって、PyMOLのダウンロードをお願いします。
大阪大学の蛋白研究所からインストールの仕方が解説されています。
簡単にダウンロードできます。
【この記事のまとめ】
「GROMACS」を使い、自宅のPCで「タンパク質-低分子リガンド複合体」の分子動力学(MD)シミュレーションを開始するための準備工程(前編)を解説した記事です。 T4リゾチームと低分子リガンド(2-プロピルフェノール)を題材に、シミュレーション実行に不可欠な「システム構築」の全手順を具体的に学べます。
- 環境構築とソフトウェアの導入: macOS環境を前提としたHomebrew経由でのGROMACSインストール方法や、構造確認のためのPyMOL準備について詳説。
- トポロジーファイルの作成: PDBデータ(3HTB)からタンパク質とリガンド(JZ4)を分離し、CHARMM36力場を用いた
pdb2gmxによる構造定義の手順を提示。 - 溶媒付加と電荷の中和: 12面体ボックス(dodecahedron)の定義、水分子の付加(
gmx solvate)、およびシステムを中性化するためのイオン置換(gmx genion)の実践フローを網羅。
この記事を読み進めることで、複雑な複合体系のMDシミュレーションにおいて最も難所とされる「前処理ステップ」を確実に完了させ、解析の土台を築くことができます。
macOS Ventura(13.2.1), PyMOL 2.5.4., Homebrew 4.0.17, Python 3.7.3, NetworkX 2.3, Avogadro, UCSF Chimera-1.17.3-mac64

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


MD シミュレーションとは?
MDシミュレーションは、分子の動きや相互作用を計算機上で再現する手法です。分子の構造と相互作用を記述する力場(potential function)を使用し、時間の経過とともに分子の運動を予測します。これにより、物質の性質や相互作用の理解、材料設計、生体分子の研究などに応用されます。
GROMACSは、MDシミュレーションを実行するためのソフトウェアの一つです。GROMACSは高性能な計算を行うための最適化が施されており、広く使われている信頼性の高いツールです。GROMACSを使うことで、分子のダイナミクスや相互作用をシミュレーションすることができます。
この記事では実際にGROMACSを使って、自分のパソコンでタンパク質(T4リゾチームL99A/M102Q)とリガンド(2-プロピルフェノール)の複合体のMDシミュレーションをを行なっていきます。
GROMACSのインストール
GROMACSを使うにあたり、Homebrewをまずターミナルからインストールします。
Homebrewは、macOS向けのパッケージ管理システムです。パッケージ管理システムは、ソフトウェアのインストールやアップデートを簡単に行えるツールであり、開発者やユーザーにとって便利です。
Homebrewを使用すると、ターミナルを介してコマンドを実行することで、様々なソフトウェアパッケージをインストールできます。これにより、開発ツール、プログラミング言語、データベース、ライブラリなど、さまざまなソフトウェアを簡単に管理できます。
Homebrewを使用すると、Mac上でGROMACSをインストールすることができます。Homebrewを使ってGROMACSをインストールすると、依存関係の解決やバージョン管理などが自動的に行われます。
まずターミナルで
/bin/bash -c "$(curl -fsSL <https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh>)"
と打ち、Homebrewをインストールします。
続いて
brew install gromacs
と打ち、gromacsをインストールします。
gmx
と打ち、以下のような記述が返ってきます。
:-) GROMACS - gmx, 2023.1-Homebrew (-:
Executable: /opt/homebrew/bin/../Cellar/gromacs/2023.1/bin/gmx
Data prefix: /opt/homebrew/bin/../Cellar/gromacs/2023.1
Working dir: /Users/kshinba
Command line:
gmx
SYNOPSIS
gmx [-[no]h] [-[no]quiet] [-[no]version] [-[no]copyright] [-nice <int>]
[-[no]backup]
OPTIONS
Other options:
-[no]h (no)
Print help and quit
-[no]quiet (no)
Do not print common startup info or quotes
-[no]version (no)
Print extended version information and quit
-[no]copyright (no)
Print copyright information on startup
-nice <int> (19)
Set the nicelevel (default depends on command)
-[no]backup (yes)
Write backups if output files exist
Additional help is available on the following topics:
commands List of available commands
selections Selection syntax and usage
To access the help, use 'gmx help <topic>'.
For help on a command, use 'gmx help <command>'.
GROMACS reminds you: "Correctomundo" (Pulp Fiction)
これが出てくるときちんとHomebrewとGROMACSのインストールが完了しています。
参考動画
MDシミュレーションは設定することが多いですが、以下の流れで進めていきます。
MD シミュレーションの流れ
- タンパク質のトポロジーファイルの準備
- リガンドのトポロジーファイルの準備
- 複合体のトポロジーファイルの準備
- ボックスを定義し、溶媒を加える
- イオンを加える (前編はここまで)
- エネルギーの最小化
- 平衡化
- MD シミュレーション
- 後処理
- MD シミュレーションの可視化
それでは順にやっていきましょう!
1. タンパク質のトポロジーファイルの準備
では今回MDシミュレーションを行うタンパク質とリガンドのトポロジーファイルの準備をしていきましょう。
トポロジーとは、特に化学や分子生物学の文脈で使用されるとき、原子や分子の結合のパターンや配置を指します。以下の順番でタンパク質とリガンドのトポロジーファイルを準備します。
- pdb2gmxというコードでタンパク質のトポロジーを準備する
- 外部ツールを使用してリガンドのトポロジーを準備する
今回はこちらのプロトコルに書いてあるタンパク質Lysozymeとそのリガンド2-propylphenolを用います。(PDBは3HTBです)
タンパク質Lysozymeは、細菌の細胞壁を分解する酵素として知られ、多くの生物において感染防御の一環として機能します。Lysozymeの活性部位は、特定のリガンドや阻害剤と結合することができるため、創薬のターゲットとして注目されています。
PDBコード3HTBには、Lysozymeとリガンド2-propylphenolの結合構造が示されています。2-propylphenolはLysozymeの活性部位に結合し、その活性を変化させる可能性があります。このようなリガンドの情報は、新しい薬物の設計や既存の薬物の改良に役立ちます。
すでに水分子とPO4を取り除いたもの(3HTB_clean.pdb)がこちらのhereからダウンロードできます。これを任意のディレクトリに保存してください。
ターミナルを開き、以下のコードでダウンロードしたディレクトリのところにいきます。
cd XXX(3HTB_clean.pdbがあるディレクトリ(ファイル))
次に以下のコードを実行して下さい。
grep JZ4 3HTB_clean.pdb > jz4.pdb
grep:テキストファイルから特定のパターンを持つ行を検索するツール。JZ4:この部分は検索したいパターンを指定しています。このコマンドでは**JZ4というパターンを持つ行を3HTB_clean.pdbファイルから検索しています。JZ4** はここではリガンドです。3HTB_clean.pdb:この部分は**grep**が検索する入力ファイルを指定しています。>:この記号はリダイレクトを意味します。コマンドの出力をファイルに保存するために使用されます。jz4.pdb:この部分は出力ファイルの名前を指定しています。このファイルには**3HTB_clean.pdbから検索されたJZ4**というパターンを持つ行の内容が保存されます。
簡単に言えば、このコマンドは**3HTB_clean.pdb**ファイルにあるタンパク質-リガンドの複合体の中から、リガンドのみのファイルを作成します。
3HTB_clean.pdb にはまだリガンドがあるので、pymolのコマンドラインから消します。以下のコードを実行して下さい。
remove resn JZ4
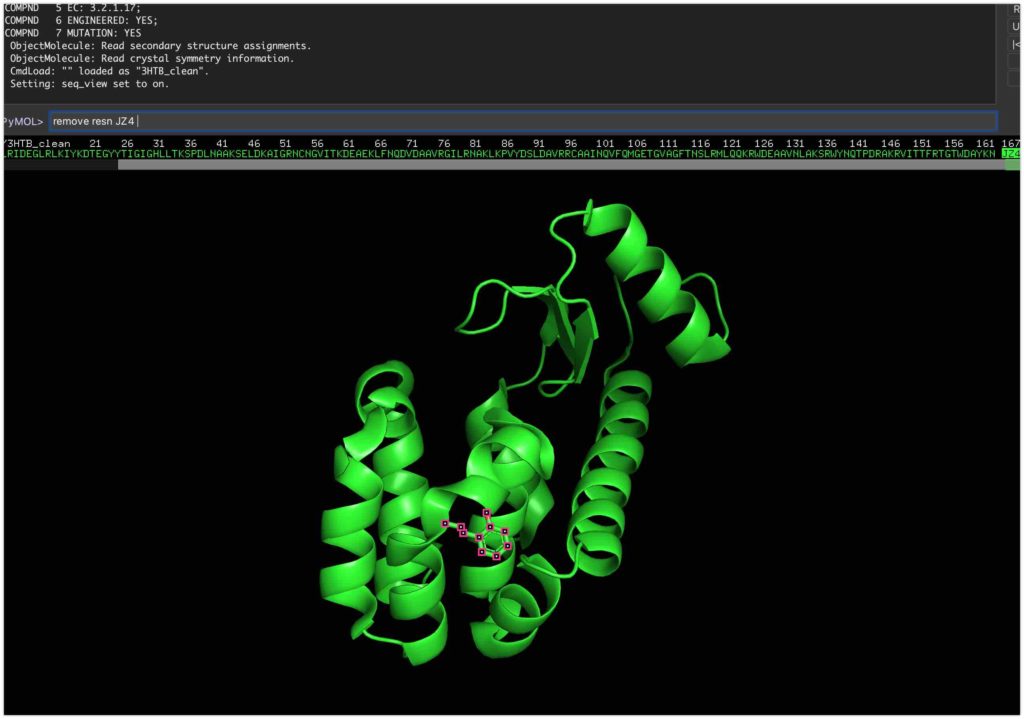
このままsaveして、リガンドが取り除かれたタンパク質Lysozymeを**3HTB_clean.pdb**として保存します。
続いてフォースフィールドの設定をします。
フォースフィールドは、分子動力学シミュレーションにおける原子間の相互作用を数学的にモデル化するためのパラメータと方程式のセットです。これには、結合の伸び、角度の変化、非結合相互作用(バンデルワールスや電気静的相互作用)などのエネルギー計算が含まれます。
このチュートリアルで使用するフォースフィールドは、MacKerell labのウェブサイトから取得したCHARMM36です。そこで、最新のCHARMM36フォースフィールドのtarballと、後で使用する”cgenff_charmm2gmx.py”変換スクリプトを現在作業しているディレクトリにダウンロードしてください。インストールされているPythonのバージョン(Python 2.xまたは3.x)に対応する変換スクリプトのバージョンをダウンロードしてください。今回、私のpythonのversionはver3で、のちに使うNetwork Xは2.3のものを使うので、cgenff_charmm2gmx_py3_nx2.pyをダウンロードします。

作業ディレクトリでフォースフィールドのtarballを解凍します:
tar -zxvf charmm36-jul2022.ff.tgz
このコマンドは、tarツールを使用して、.tgz(または**.tar.gz)形式の圧縮アーカイブファイルを展開するものです。具体的には、charmm36-jul2022.ff.tgz**という名前のファイルを展開します。
コマンドのオプションは以下のように解釈されます:
z:.gz(gzip)形式で圧縮されたアーカイブの展開を指示します。x: アーカイブを展開するモードで**tar**を実行します。v: 展開されるファイルの名前を表示します(verboseモード)。f: 続く引数(この場合は**charmm36-jul2022.ff.tgz**)をアーカイブファイル名として解釈します。
したがって、**tar -zxvf charmm36-jul2022.ff.tgzというコマンドを実行すると、charmm36-jul2022.ff.tgz**という圧縮アーカイブが現在のディレクトリに展開され、展開されるファイルの名前が表示されます。
作業ディレクトリに”charmm36-jul2022.ff”サブディレクトリがあるはずです。
以下のコードでpdb2gmxでT4リゾチームのトポロジーを記述します:
gmx pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro -ter
**gmx pdb2gmx**は、GROMACSツールの一部として提供されているコマンドで、PDBフォーマットのタンパク質構造をGROMACSが理解できる形式に変換するためのものです。
以下は、指定されたコマンドの各オプションとその説明です:
f 3HTB_clean.pdb:入力として使用するPDBファイルの名前を指定します。この例では、**3HTB_clean.pdb**という名前のファイルを入力として使用します。o 3HTB_processed.gro:出力ファイルの名前を指定します。変換が完了すると、この名前でGROMACSの.gro形式のファイルが生成されます。ter:タンパク質のC-末端とN-末端について、ターミナルグループの選択をユーザーに問い合わせるオプション。タンパク質の末端にはいくつかの異なるターミナルグループが選択できるため、このオプションはユーザーに選択の機会を与えるものです。
このコマンドを実行すると、GROMACSは入力として提供されたPDBファイルを読み取り、それを基にGROMACS用の構造ファイルを生成します。このプロセス中に、タンパク質の末端のタイプ、欠損している原子の処理、使用する力場の選択など、いくつかのユーザー入力が求められます。
3つの選択をするように求められます。
- フォースフィールド
- 水モデル
- 端のタイプ
まず以下のように出てきます。”1”を選択し、CHARMMフォースフィールドを選択します。
Select the Force Field:
From current directory:
1: CHARMM all-atom force field
From '/opt/homebrew/bin/../Cellar/gromacs/2023.2/share/gromacs/top':
2: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
3: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
4: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
5: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
6: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
7: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
8: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
9: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
10: GROMOS96 43a1 force field
11: GROMOS96 43a2 force field (improved alkane dihedrals)
12: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
13: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
15: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
16: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
CHARMM(Chemistry at HARvard Macromolecular Mechanics)フォースフィールドは、分子動力学シミュレーションで使用される一連のパラメータと数学的方程式のセットであり、分子の挙動を計算するためのものです。フォースフィールドは、原子間の相互作用や結合、角度、二面角などのエネルギーを計算するためのパラメータを提供します。
CHARMMフォースフィールドの特徴:
- 多様性: CHARMMは、多くの異なる生物分子や有機分子に適用できるように設計されており、これによりタンパク質、核酸、炭水化物、脂質二重層などのシミュレーションが可能です。
- 周期的更新: CHARMMフォースフィールドは、新しい実験的データや計算結果に基づいて定期的に更新され、精度を向上させています。
- 適用範囲: 有機分子や生物分子、金属イオンとの相互作用など、幅広い化学的環境に適用可能です。
- エネルギー項: CHARMMフォースフィールドは、結合エネルギー、結合角エネルギー、二面角エネルギー、非結合エネルギー(ファンデルワールス相互作用と電荷-電荷相互作用)を計算するための数学的表現を提供します。
フォースフィールドは、分子動力学シミュレーションの精度と効率性を決定する重要な要因であり、適切なフォースフィールドの選択は、シミュレーション結果の信頼性を確保するために重要です。CHARMMは、その詳細なパラメータ化と幅広い適用範囲により、多くの研究者によって広く使用されているフォースフィールドの一つです。
以下のように出てきます。”1”をターミナルで記述しEnterを押します。
Select the Water Model:
1: TIP3P CHARMM-modified TIP3P water model (recommended over original TIP3P)
2: TIP3P_ORIGINAL Original TIP3P water model
3: SPC SPC water model
4: SPCE SPC/E water model
5: TIP5P TIP5P water model
6: TIP4P TIP4P water model
7: TIP4PEW TIP4P/Ew water model
8: None
ここで選択したTIP3Pは、水分子をモデル化するための広く使用されている3点荷電モデルの一つです。このモデルでは、水分子は2つの水素原子と1つの酸素原子から構成され、酸素には部分的な負の電荷が、各水素には部分的な正の電荷が存在します。また、酸素と水素間の結合と、2つの水素原子間の角度に関する構造パラメータも定義されています。
“CHARMM-modified TIP3P”は、TIP3Pモデルの改良版で、CHARMMフォースフィールド内での使用を意図して最適化されています。この改良は、CHARMMの他の成分(例えば、タンパク質や脂質)と相互作用する水の動きやエネルギーの挙動をより正確に再現するためのものです。
一般に、特定のフォースフィールドのコンテキスト内で最適な結果を得るためには、そのフォースフィールドのために特別に設計または改良された水モデルを使用することが推奨されます。この場合、CHARMMフォースフィールドでの使用を前提としたTIP3Pの修正版が推奨されており、元のTIP3Pよりもこの修正版を使用する方が好ましいとされています。
以下のように出てきます。”1”をターミナルで記述しEnterを押します。
Special Atom Distance matrix:
MET1 MET6 HIS31 CYS54 CYS97 MET106
SD10 SD56 NE2276 SG446 SG808 SD883
MET6 SD56 0.631
HIS31 NE2276 2.196 1.931
CYS54 SG446 2.781 2.718 1.052
CYS97 SG808 0.820 0.539 2.117 2.897
MET106 SD883 1.900 1.314 1.848 2.841 1.635
MET120 SD982 2.525 2.014 3.286 4.293 2.112 1.520
Select start terminus type for MET-1
0: MET1
1: NH3+
2: NH2
3: HYD1
4: 5TER
5: 5MET
6: 5PHO
7: 5POM
8: None
次のように出てきます。”0”をターミナルで記述しEnterを押します。
Start terminus MET-1: NH3+
Select end terminus type for ASN-163
0: COO-
1: COOH
2: CT2
3: CT1
4: HYD2
5: MET2
6: 3TER
7: None
これらはN末端とC末端を定義しています。
以下のようなメッセージが出たら、タンパク質のトポロジーファイルの作成に成功しています。
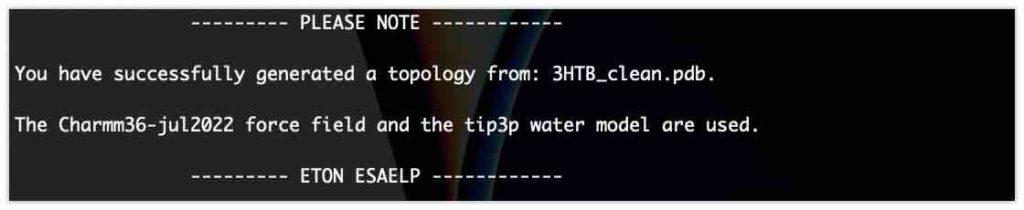
これにてタンパク質のトポロジーファイルの作成は終了です。
リガンドのトポロジーファイルの準備
続いて、リガンドのトポロジーファイルの準備をAvogadroとCGenFFを駆使して行います。
まずAvogadroをダウンロードして下さい。
Avogadroでjz4.pdbを開き、上のビルドタブから水素を追加して下さい。
File -> Save As… から Sybyl Mol2を選択し、jz4.mol2として保存。
保存したjz4.mol2をテキストファイルで開き、上部の*****の箇所をJZ4に置き換えます。
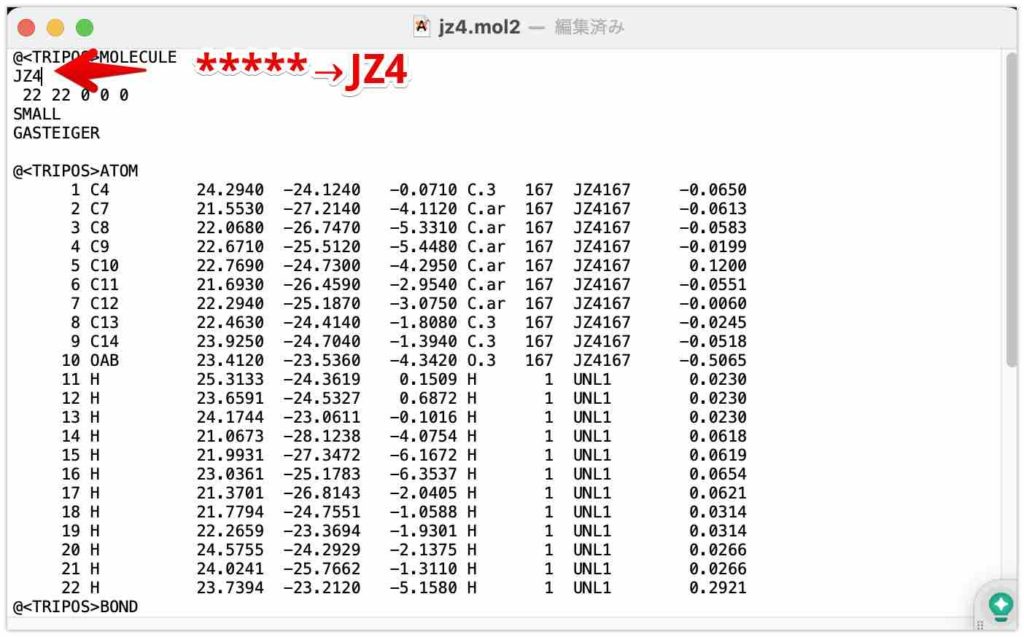
今の状態ではjz4.mol2 の@<TRIPOS>BONDのセクションの順序を変更する必要があるそうです。次のサイトから以下のsort_mol2_bonds.plファイルをダウンロードして下さい。

Macでは新しいタブが開かれるだけなので、これをVisual Studio codeなどにコピペして、同じディレクトリに保存して下さい。以下が参考動画です。
続いて、ターミナルで以下のコードを実行して下さい。
perl sort_mol2_bonds.pl jz4.mol2 jz4_fix.mol2
これにより、jz4_fix.mol2 が新しく生成されることを確認して下さい。
このファイルを使って、リガンドのJZ4トポロジーをCGenFFというツールで生成します。
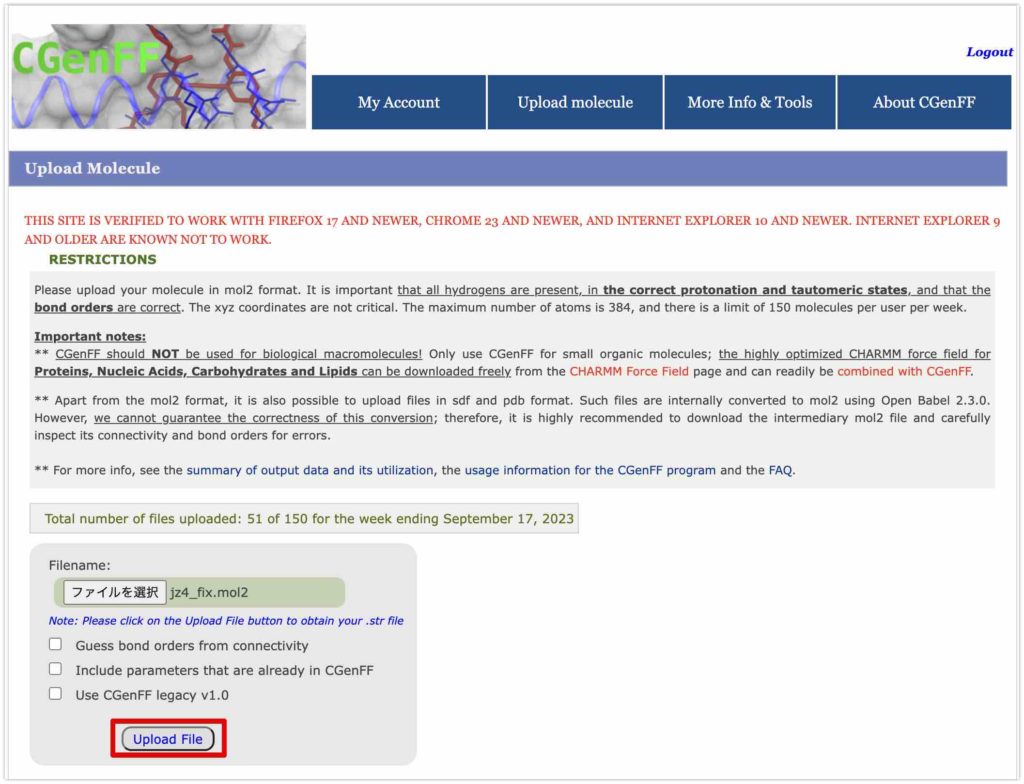
続いて、CGenFFのサイトにいき、アカウント登録をして下さい。
CGenFFに生成したjz4_fix.mol2 ファイルをアップロードします。
jz4.strが生成されます。
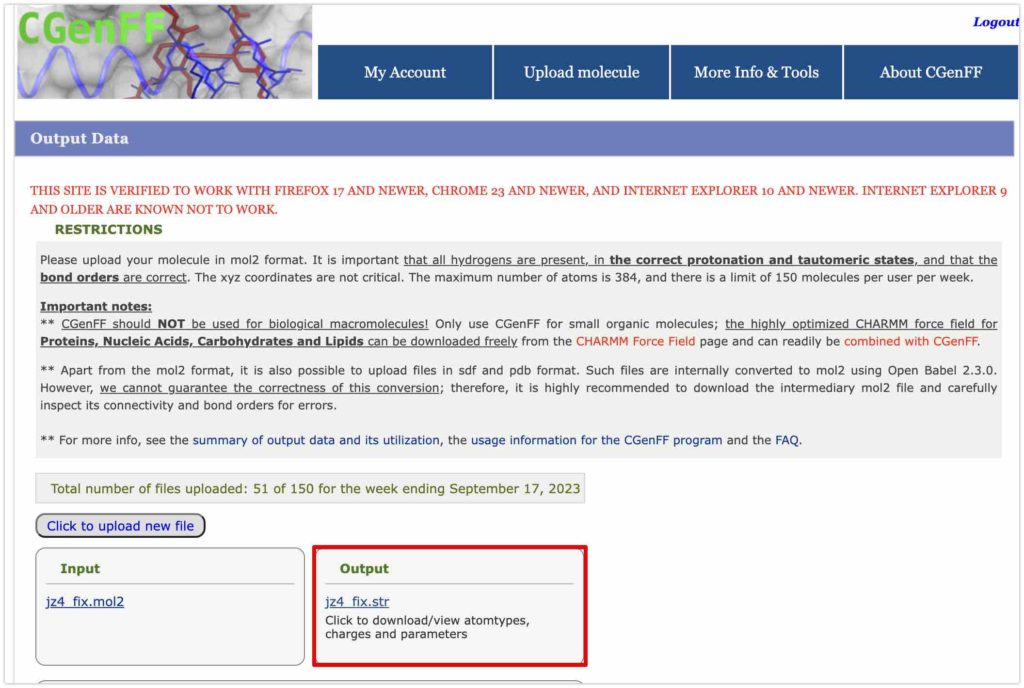
こちらをクリックすると新しくタブが開きます。
これをコピペしておきます。
terminalで以下を実行してください。
nano jz4.str
その後、先ほどコピペしたものを貼り付けて ctrl+X→Yを押してください。
続いて、以下のコードを打ち、現在のディレクトリでpython ver 3.5.2もしくは3.7.3を実行する環境を整えます。環境はAnacondaを使っています。**my_new_env**という名前の新しいAnaconda環境を作成し、その環境にPython 3.7.3をインストールします。
conda create --name my_new_env python=3.7.3
以下のコードで新しい環境をアクティベートします。
conda activate my_new_env
続いて以下のコードでNetworkX 2.3のインポートして下さい。
pip install networkx==2.3
以下のコードを実行して下さい。
python cgenff_charmm2gmx_py3_nx2.py JZ4 jz4_fix.mol2 jz4.str charmm36-jul2022.ff
※ここでは適切なpythonの3.5.2 versionとNetworkX 2.3 設定されていないと上手くスクリプトが動かないので、注意してください。以下のように出ると成功しています。
============ DONE ============
Conversion complete.
The molecule topology has been written to jz4.itp
Additional parameters needed by the molecule are written to jz4.prm, which needs to be included in the system .top
PLEASE NOTE: If your topology has lone pairs, you must use GROMACS version 2020 or newer to use 2fd construction
Older GROMACS versions WILL NOT WORK as they do not support 2fd virtual site construction
============ DONE ============
※私の場合、ダウンロードしたcgenff_charmm2gmx_py3_nx2.pyでは上手くいかなかったため、
以下の部分を
elif section == "DIHE":
if line.find('!'):
line = line[:line.find('!')]
s = line.split()
ai, aj, ak, al, k, n, d = s[0],s[1],s[2],s[3],float(s[4]),int(s[5]),float(s[6])
parameters["DIHE"].append([ai,aj,ak,al,k,n,d])
次のように変更しました。
elif section == "DIHE":
line = line.split('!')[0] # コメント部分を除去
s = line.split() # スペースでデータ項目を分割
if len(s) < 7:
continue # 7つの要素がない場合は、次の行に進む
ai, aj, ak, al = s[0], s[1], s[2], s[3]
k = float(s[4])
n = int(s[5])
d = float(s[6])
parameters["DIHE"].append([ai,aj,ak,al,k,n,d])
続いて以下のコマンドを実行してください。
gmx editconf -f jz4_ini.pdb -o jz4.gro
gmx:- GROMACSの主要なコマンド実行ツール。これに続けて使用するツールやモジュール名(この場合は
editconf)を指定します。
- GROMACSの主要なコマンド実行ツール。これに続けて使用するツールやモジュール名(この場合は
editconf:- GROMACSに内包されている、構造ファイルを編集するためのツールです。このツールを使用すると、入力ファイルの形式を変換したり、シミュレーションボックスの形状やサイズを変更したり、他の多くのタスクを実行することができます。
f jz4_ini.pdb:fオプションは「input file」を意味します。この後に続くjz4_ini.pdbは入力として使用するファイルの名前です。この場合、PDB形式のjz4_ini.pdbファイルを編集対象として指定しています。
o jz4.gro:oオプションは「output file」を意味します。この後に続くjz4.groは出力ファイルの名前です。このコマンドの実行後には、入力ファイルの内容が.gro形式でjz4.groという名前のファイルに保存されます。
簡単に言うと、このコマンドは jz4_ini.pdb という名前の PDB ファイルを読み込み、それを GROMACS の .gro 形式に変換して、jz4.gro という名前の新しいファイルに保存する操作を行います。
これにてリガンドのトポロジーファイルの準備は終了です。
複合体のトポロジーファイルの準備
続いてタンパク質とリガンドの複合体ファイルを作っていきます。
3HTB_processed.groをコピーして新しくcomplex.groというファイルを作ってください。このcomplex.gro を修正して複合体のファイルを作ります。
ファイルを開き、complex.groの下部にリガンドのJZ4のものを貼り付けます。
complex.groの下部に
163ASN C 1691 0.621 -0.740 -0.126
163ASN O1 1692 0.624 -0.616 -0.140
163ASN O2 1693 0.683 -0.703 -0.011
5.99500 5.19182 9.66100 0.00000 0.00000 -2.99750 0.00000 0.00000 0.00000
赤字を付け加えてください。
163ASN C 1691 0.621 -0.740 -0.126
163ASN O1 1692 0.624 -0.616 -0.140
163ASN O2 1693 0.683 -0.703 -0.011
1JZ4 C4 1 2.429 -2.412 -0.007
1JZ4 C7 2 2.155 -2.721 -0.411
1JZ4 C8 3 2.207 -2.675 -0.533
1JZ4 C9 4 2.267 -2.551 -0.545
1JZ4 C10 5 2.277 -2.473 -0.430
1JZ4 C11 6 2.169 -2.646 -0.295
1JZ4 C12 7 2.229 -2.519 -0.308
1JZ4 C13 8 2.246 -2.441 -0.181
1JZ4 C14 9 2.392 -2.470 -0.139
1JZ4 OAB 10 2.341 -2.354 -0.434
1JZ4 H1 11 2.531 -2.436 0.015
1JZ4 H2 12 2.366 -2.453 0.069
1JZ4 H3 13 2.417 -2.306 -0.010
1JZ4 H4 14 2.107 -2.812 -0.407
1JZ4 H5 15 2.199 -2.735 -0.617
1JZ4 H6 16 2.304 -2.518 -0.635
1JZ4 H7 17 2.137 -2.681 -0.204
1JZ4 H8 18 2.178 -2.476 -0.106
1JZ4 H9 19 2.227 -2.337 -0.193
1JZ4 H10 20 2.458 -2.429 -0.214
1JZ4 H11 21 2.402 -2.577 -0.131
1JZ4 H12 22 2.374 -2.321 -0.516
5.99500 5.19182 9.66100 0.00000 0.00000 -2.99750 0.00000 0.00000 0.00000
一番上の番号を2636にします。

続いて複合体のトポロジーファイルを作っていきます。
テキストファイルでtopol.topを開く。赤字を付け加えてください。
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
; Include ligand topology
#include "jz4.itp"
; Include water topology
#include "./charmm36-jul2022.ff/tip3p.itp"
またtopol.topに以下の赤字も付け加えてください。
; Include forcefield parameters
#include "./charmm36-jul2022.ff/forcefield.itp"
; Include ligand parameters
#include "jz4.prm"
[ moleculetype ]
; Name nrexcl
Protein_chain_A 3
[ molecules ]
; Compound #mols
Protein_chain_A 1
JZ4 1
以上で複合体のトポロジーファイルの生成は終了です。
ボックスを定義し、溶媒を加える
続いてMD シミュレーションの箱を定義し、その中に溶媒を加えます。
以下のコードを実行してください。
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0
gmx:- GROMACSの主要なコマンド実行ツールです。
editconf:- GROMACSの構造ファイル編集ツール。
f complex.gro:- 入力として使用するファイルの名前を指定するオプション。この場合、
complex.groという名前の GROMACS 形式のファイルを使用します。
- 入力として使用するファイルの名前を指定するオプション。この場合、
o newbox.gro:- 出力ファイルの名前を指定するオプション。このコマンドの結果は、
newbox.groという名前のファイルに保存されます。
- 出力ファイルの名前を指定するオプション。このコマンドの結果は、
bt dodecahedron:btオプションはシミュレーションボックスのタイプを指定します。dodecahedronは12面体の形状のシミュレーションボックスを意味します。12面体のボックスは、球形のタンパク質や分子の周りに余分な溶媒を最小限に抑えるのに役立つ形状として知られています。
d 1.0:dオプションは、シミュレーションボックスの壁と最も近い分子の間の最小距離を指定します。この場合、1.0 nm (ナノメートル) が指定されています。この距離を設定することで、分子がボックスの境界に接触することなく、自由に動くことが確保されます。
このコマンドは complex.gro ファイルを読み取り、シミュレーションボックスを12面体の形状に変更し、分子とボックスの壁との間の最小距離を1.0 nmに設定して、その結果を newbox.gro ファイルに保存します。
続いて以下のコードを実行してください。
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
gmx solvate: GROMACSのツールで、分子を溶媒中に配置するためのものです。cp newbox.gro: このオプションは、溶媒を追加する前の初期構造のファイルを指定します。この例では**newbox.gro**がそのファイルです。cs spc216.gro: 使用する溶媒モデルを指定します。**spc216.gro**はSPCモデルの水分子を含む一般的なファイルで、多くのシミュレーションで使用される溶媒です。p topol.top: トポロジーファイルを指定します。このファイルには、システムの各分子のトポロジー情報(結合、角度、二面角など)が含まれています。溶媒を追加すると、このファイルも更新され、追加された水分子の数が反映されます。o solv.gro: 出力ファイルの名前を指定します。この例では、溶媒が追加された後の新しい構造が**solv.gro**という名前のファイルに保存されます。
このコマンドを実行することで、**newbox.groに指定された構造をspc216.groの水分子で溶解し、その結果をsolv.groという名前の新しいファイルに保存します。また、topol.top**トポロジーファイルも更新され、システムに追加された水分子の数が反映されます。
これにてボックスを定義し、溶媒を加える作業は終了です。
イオンを加える
続いてイオンを加えていきます。
以下のコードを実行してください。
nano ions.mdp
これを実行すると、**ions.mdpというファイルがnanoエディタ内で開かれます。もしions.mdp**というファイルが存在しない場合、新しく空のファイルが開かれます。
このページをコピぺしてください。元データはこちらから入手できます。
ctrl X→Y→Enterを押して保存してください。
続いて次のコードを実行してください。
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr
gmx grompp: GROMACSのユーティリティで、シミュレーションの前処理ステップを行います。f ions.mdp: シミュレーションのパラメータを定義する**.mdpファイルを指定します。この例では、ions.mdp**というファイルがそのファイルです。c solv.gro: 初期構造を指定します。**solv.gro**は溶媒を含むシステムの初期構造を表します。p topol.top: トポロジーファイルを指定します。**topol.top**にはシステムの各分子のトポロジー情報(結合、角度、二面角など)や使用されている力場の情報が含まれています。o ions.tpr: 出力ファイルの名前を指定します。このユーティリティの実行により、**ions.tprという名前のバイナリ入力ファイルが生成されます。この.tpr**ファイルは、次のステップであるシミュレーションの実行に使用されます。
このコマンドを実行すると、指定された構造、トポロジー、およびシミュレーションパラメータを使用して、シミュレーションの入力ファイルが作成されます。このステップが成功すると、次に**gmx mdrun**などのコマンドを使用して実際のシミュレーションを実行できます。
続いて以下のコードを実行してください。
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral
gmx genion: シミュレーションボックスにイオンを追加するためのGROMACSユーティリティ。s ions.tpr: イオンを追加する前のシステムのバイナリ入力ファイル。このファイルは、前のステップ(gmx grompp)で生成されたものです。o solv_ions.gro: イオンを追加した後のシステムの構造ファイルの名前。p topol.top: システムのトポロジーファイル。このファイルは、イオンが追加されることで更新されます。pname NA: 正のイオンの名前(この場合はナトリウム、Na^+)。nname CL: 負のイオンの名前(この場合は塩化物、Cl^-)。neutral: このオプションを使用すると、システム全体の電荷が中和されるようにイオンが追加されます。つまり、正と負のイオンが適切な比率で追加され、全体の電荷がゼロになります。
このコマンドを実行すると、通常、水分子の一部が指定されたイオンに置き換えられます。この置換は、シミュレーションボックスの電荷を中和するため、または指定されたイオン濃度にするために行われます。
実行すると以下のように出てくるかと思います。
Select a continuous group of solvent molecules
Group 0 ( System) has 33518 elements
Group 1 ( Protein) has 2614 elements
Group 2 ( Protein-H) has 1301 elements
Group 3 ( C-alpha) has 163 elements
Group 4 ( Backbone) has 489 elements
Group 5 ( MainChain) has 651 elements
Group 6 ( MainChain+Cb) has 803 elements
Group 7 ( MainChain+H) has 813 elements
Group 8 ( SideChain) has 1801 elements
Group 9 ( SideChain-H) has 650 elements
Group 10 ( Prot-Masses) has 2614 elements
Group 11 ( non-Protein) has 30904 elements
Group 12 ( Other) has 22 elements
Group 13 ( JZ4) has 22 elements
Group 14 ( Water) has 30882 elements
Group 15 ( SOL) has 30882 elements
Group 16 ( non-Water) has 2636 elements
溶媒は多くの場合、SOLが一般的だそうです。そのため、15を選びます。
solv_ions.gro ファイルが出来ていると、完成しています。
またtopol.topファイルの一番下が以下のようになっていれば大丈夫です。
[ molecules ]
; Compound #mols
Protein_chain_A 1
JZ4 1
SOL 10228
CL 6
お疲れ様でした。一旦前編はこちらで終了です。後編はこちらから。後編からは実際にシミュレーションをやっていきます。
参考資料
Molecular Dynamics Tutorial | Protein-Ligand Complex: GROMACS – PART 1
Molecular Dynamics Tutorial | Protein-Ligand Complex: GROMACS – PART 2
EP 11 | Post MD Simulation movie for visualization Protein-Ligand complex trajectories | Gromacs
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

