
本記事はAutodock Vinaを使ったin silico薬物スクリーニングついて書かれた記事です。in silico薬物スクリーニングは膨大な数の小さな分子からリード化合物を特定するための強力なツールとなっています。本記事では実際に糖尿病の原因タンパク質に対して、実際にin silicoスクリーニングを行い、薬物候補化合物を取ってきます。自分のパソコンで簡単にできるので、ぜひトライしてみてください!
Parallel desktopで構築したWindows 11 Pro (22H2). PC自体はmacOS Ventura(13.2.1)
(通常のwindowsで大丈夫です)
OpenBabelGUI(2.3.1)、 MGLTools(1.5.7) MGLToolsがMacでは使えません!
perl 5, version 14, subversion 2 (v5.14.2) built for MSWin32-x86-multi-thread
※Parallel desktopは課金しますが、比較的安く(1万円/年)、簡単にMac上にwindows環境を構築することができます。
興味ある方はこちらから試してみてください。面倒な仮想環境を構築をすることなく、普通のアプリのようにwindowsが試せるので、結構おすすめです。

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1500 今なら50%OFF!!
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています

自宅でできるin silico創薬の技術書を販売中
¥3,200 → ¥1,500 今なら50%OFF!!
分子ドッキングやMDシミュレーションなど、
自宅でできるin silico創薬の解析方法を解説したものになります!
AutoDock Vinaとは?
AutoDock Vinaは、分子ドッキングとバーチャルスクリーニングのためのオープンソースソフトウェアです。主な利点は以下の通りです:
- 高速性: 最適化されたアルゴリズムにより迅速な計算が可能。
- 精度: AutoDockの改良版として、高いドッキング精度を持つ。
- 使いやすさ: シンプルなコマンドラインインターフェースを採用。
- 柔軟性: 多様なリガンドとターゲットタンパク質に適応。
- 無料: オープンソースであり、カスタマイズが可能。
- 大規模スクリーニング: 大量の化合物ライブラリを効率的に処理。
- マルチスレッド対応: 複数のCPUコアでの高速化が可能。
これらの特徴により、Vinaはin silicoスクリーニングの強力なツールとして利用されています。
In silico screeningとは?
In silico screening(インシリコスクリーニング)とは、バーチャルスクリーニングの一種で、コンピューターシミュレーションを使用して、大規模な分子ライブラリーから、特定の生物学的標的に対して有望な化合物を選別するプロセスです。
“In silico”とは、ラテン語の「in silicium(シリコン中)」に由来し、コンピューター上でのシミュレーションを意味しています。
In silico screeningでは、コンピューターモデルを用いて、大量の化合物の構造情報や生物学的活性を予測し、検討対象とするターゲットに対して最適な化合物を探索することができます。
このプロセスは、実験的に合成する前に、有望な化合物を選別し、合成の労力や費用を節約することができます。また、実験的なスクリーニングよりも高速であり、より多くの化合物を同時に検討できるため、薬物開発の効率を高めることができます。
In silico screeningは、薬剤の探索や、化学物質の特性予測、新しい材料の設計など、様々な分野で活用されています。
今回はパソコンのスペック上、大規模ライブラリーを構築することは難しいので、3つの化合物からなる小規模ライブラリを使ってin silico screeningを行います。
では早速AutoDock Vinaを使ったin silico screeningを試してみましょう!
手順としては以下のように行います。
- 標的タンパク質の準備
- 化合物ライブラリの準備
- in silicoスクリーニングの設定
- in silicoスクリーニング
標的タンパク質の準備
続いて、in silicoスクリーニングの標的の下準備をしていきます。Protein Data Bankのタンパク質の多くはそのバインダーとの複合体なので、それを取り除いていきます。今回は糖尿病の標的である、DPP4(pdb:2OQV)を例にして解説します。
まずpymolを開き、
FIle→GetPDB…をクリックし、PDB IDに2OQVを入れ、downloadを押し、pymol上にDPP4を表示させます。
続いてDPP4は二つの同じユニットの構成されるタンパク質なので、片方のユニットだけを指定します。
pymolのコマンドラインに
sele chain A
と記入し実行してください。sele chain Aとは、鎖IDがAである分子を選択するコマンドです。
また
color red, sele
と実行して、分かりやすいようにchain Aの色を赤くします。
Chain Aにはバインダーが含まれているので、これを消去します。
上のDisplay→Sequenceから配列を表示させ、一番右にあるMA9を選択し、
sele→remove atomsからバインダーを消去します。
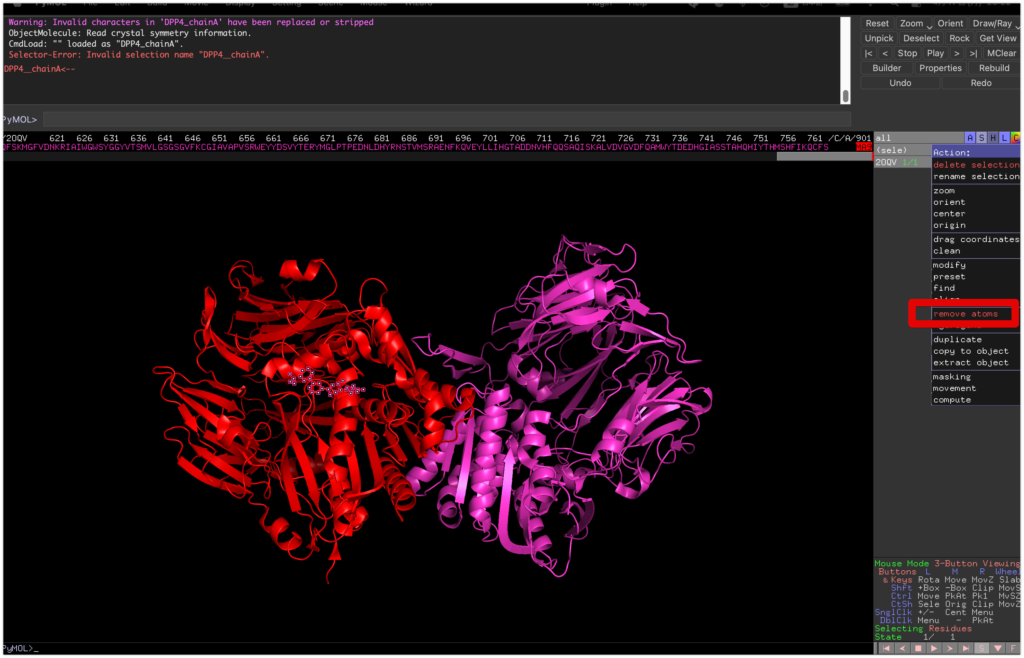
そして、再度sele chain Aからchain Aを選択し、
save DPP4_prep.pdb, sele
からDPP4_prep.pdbという名前でchain AをPDBファイルとして保存します。
続いて、MGLToolsを使って設定を行っていきます。
MGLToolsは、分子のモデリングやビジュアル化をサポートする一連のプログラムを含むソフトウェアスイートです。中でも、AutoDockの前処理や結果の解析・視覚化に使用されるツールが含まれています。AutoDockは、タンパク質とリガンドの分子間の結合予測を行うための無料のドッキングソフトウェアであり、MGLToolsはその入力ファイルの作成や出力の解析に不可欠です。したがって、MGLToolsとAutoDockは密接に連携しており、複合体の予測や薬物設計の研究において一緒に使用されることが多いです。
MGLToolを開き、左上のFile→Read Moleculeから先ほど作ったDPP4_prep.pdbを開いてください。
Grid→Macromolecule→chose→DPP4_prepを選択し、DPP4_prepをAutoDock用のpdbqtファイルに保存します。
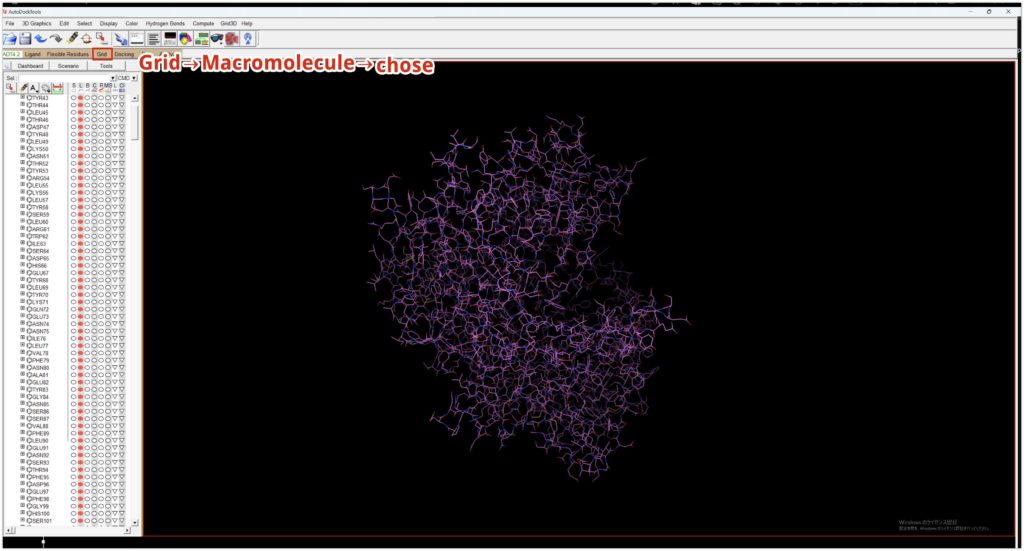
続いて、リガンドが結合するサイトを決めていきます。
先ほどの保存したDPP4_prep.pdbqtを開き、Grid→Gridboxを開くと、ボックスが出てきます。
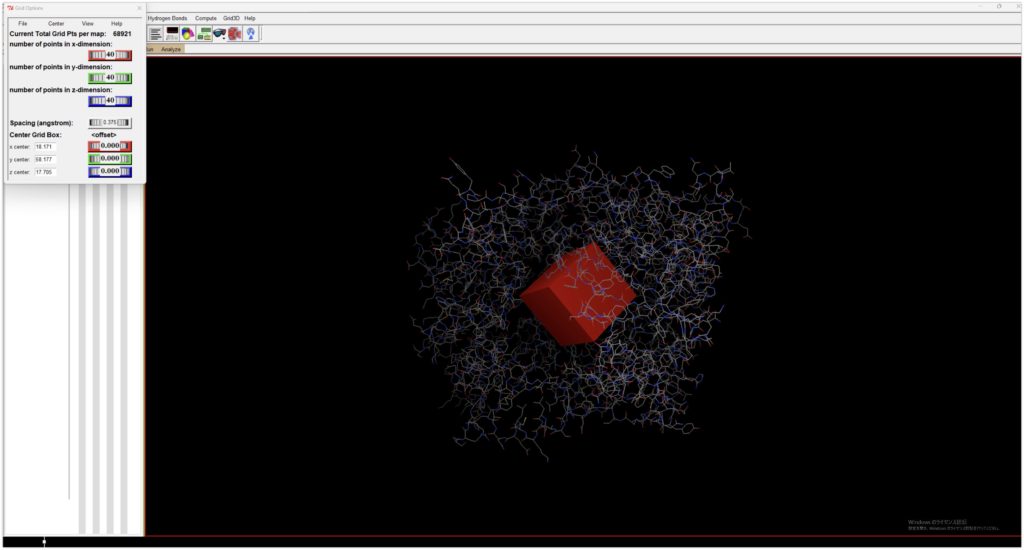
今回は結合サイトがわからないので、できるたけこのボックスを大きくします。number of points in x-dimensionのところなどをドラッグして、タンパク質がこのボックスに入るようにしてください。(以下の図では最大値の126としていますが、メモリの関係で80が丁度よさそうです。)
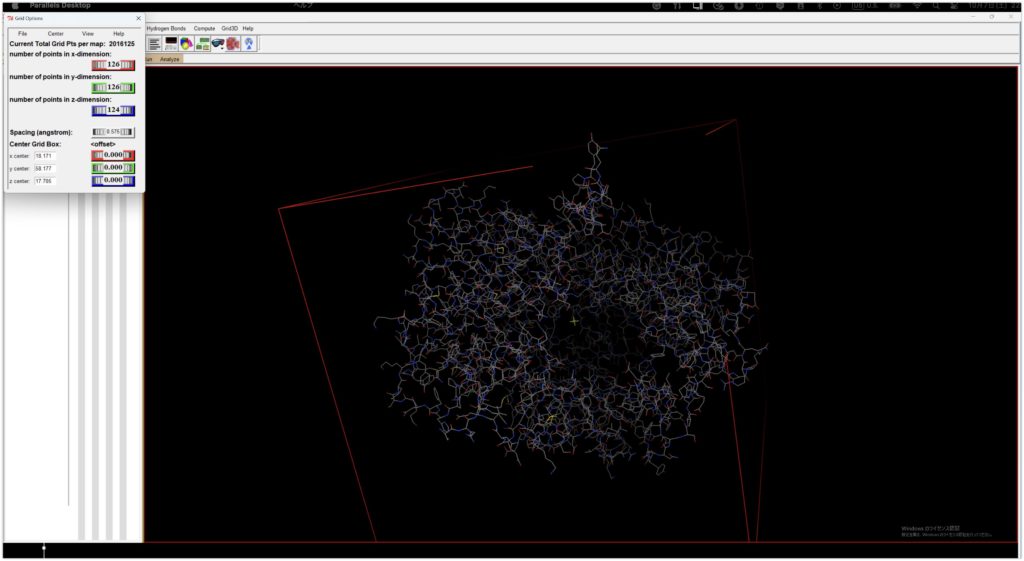
大体こんな感じです。わかりやすいようにView→show boxes as linesにしてボックスを線でわかるようにしています。
調節したGrid Boxの情報はのちに使うので、Grid OptionのFile→Output grid dimensions Fileとして保存しておきます。
これにてタンパク質の準備は終了です。
ZINCデータベースからの化合物ライブラリの構築
ZINCデータベースは、化学化合物の仮想スクリーニングに使用される公開された化学データベースの一つです。化学化合物のデータや情報を提供し、新薬の探索や設計に役立ちます。
ZINCデータベースは、様々な化学化合物のデータを含んでおり、化学構造、物性、生物活性、参考文献などが含まれています。これにより、研究者や薬剤師は、様々な化学化合物の情報を簡単に検索し、必要なデータを入手することができます。
ZINCデータベースは、主に仮想スクリーニングという手法に使用されます。仮想スクリーニングは、コンピュータ上で膨大な数の化学化合物を高速にスクリーニングし、有望な化合物を選択する手法です。ZINCデータベースは、このような仮想スクリーニングに利用されることが多く、新薬の探索や設計を支援するための有用なツールとして広く使用されています。
ここでは簡単な化合物ライブラリとして、Omarigliptin、Anagliptin、ZINC33の小さな三つのライブラリを作りましょう。
ZINCデータベースのサイトにアクセスし、上記タブのSubstancesを押してください。
ここから望みの低分子化合物を取得できます。
ここではすでにDPP4に対して作用があることが知られているOmarigliptinを例にして説明します。
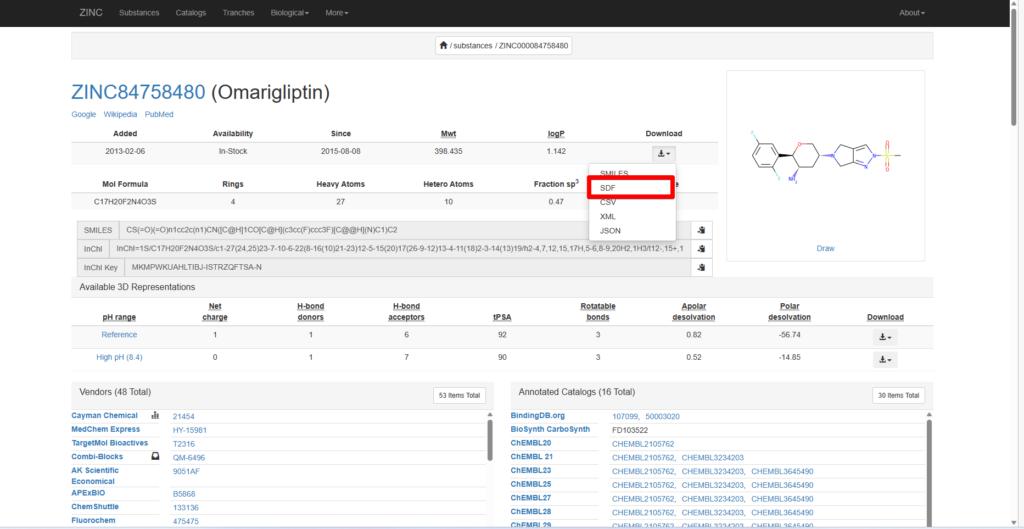
SearchバーよりOmarigliptinを検索し、Omarigliptinを選択すると以下のページが出てきます。
ここからダウンロードを押し、SDFファイルを保存してください。
同様にして、すでにDPP4に対して作用があることが知られているAnagliptin、全く関係のない化合物ZINC33についても行います。
続いて、SDFファイルをPDBファイルへと変換します。これには一般的にOpen Babelを使います。
Open Babelは、化学情報の変換と処理を行うオープンソースのツールキットです。100以上の化学データフォーマットをサポートし、異なるソフトウェアやデータベース間でのデータの互換性を提供します。主に、化学構造のフォーマット変換に使用されます。簡単に言えば、化学情報の「翻訳者」として機能します。
こちらからOpen Babelの2.3.1をダウンロードしてください。
※最新の3.1.1についてはなぜかこの後の出力形式であるpdbqtが使えませんでした。。。
そのあと、OpenBabelを開き、inputファイルにリガンドのsdfファイルを指定してください。
OUTPUT FORMATにはpdbqtを指定してください。
Add hydrogensにチェックを入れてください。(チェックを入れたものの、きちんと水素がすべての炭素などについていないことがあります。注意してください)
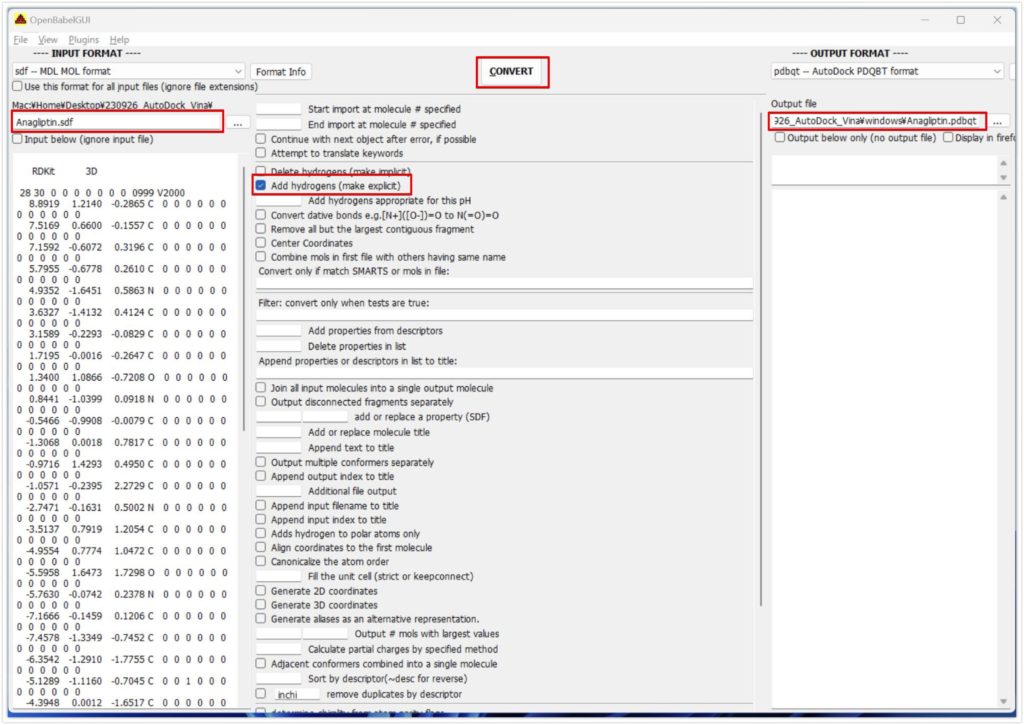
最後にConvertを押すと、pdbqtファイルが出てきます。これをすべてのリガンドに対して行って下さい。
これにてライブラリの下準備は終了です。
In silico screeningの設定
次にこちらからAutoDock vinaをダウンロードしてください。
ダウンロードしたAutoDock vinaにあるファイル(C:\Program Files (x86)\The Scripps Research Institute\Vinaにあると思います。)にあるvinaとvina_licenseとvina_splitのファイルを移動させて、
タンパク質があるファイルに入れます。ここではそのファイルをVina Dockingとしています。
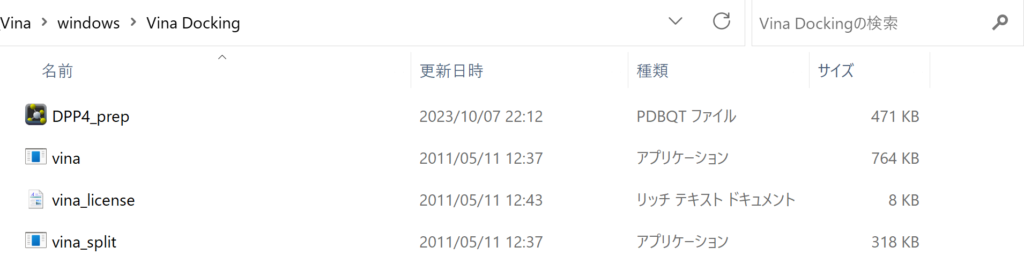
続いて、こちらからperlファイルも入れます。このファイルも同じディレクトリに入れておいてください。また、すべてのリガンドもこのファイルに入れてください。
そして、新たにconf_vsというtxtファイルを作成してください。
その中に次の文章を入れてください。receptorについては.pdbqtと拡張子がないとうまく動かないので、注意してください。
receotor = DPP4_prep.pdbqt
center_x = 18.171
center_y = 58.177
center_z = 17.705
size_x = 80
size_y = 80
size_z = 80
num_modes = 10
energy_range = 4
conf_vs.txt ファイルは、AutoDock Vinaを使用してドッキング計算を行うための設定ファイルとして見られます。以下は、提供されたファイルの各項目の説明です:
- receptor:
DPP4_prep.pdbqtはドッキングに使用されるレセプターのファイル名を指定しています。レセプターは、リガンドが結合するタンパク質または他の大きな分子を指します。.pdbqtフォーマットは、AutoDockやAutoDock Vinaで使用される特別なフォーマットであり、原子の座標の他に、原子タイプや部分的な電荷情報を持っています。
- center_x, center_y, center_z:
- これらは探索空間の中心の座標を指定しています。探索空間は、リガンドがどの範囲でタンパク質との間に結合を試みるかを示しています。この座標は通常、ドッキングの注目領域や既知の結合サイトの中心に設定されます。
- size_x, size_y, size_z:
- これらは探索空間のサイズを指定しています。各方向(X, Y, Z)での探索空間のサイズをアングストローム単位で示しています。このサイズは、探索を行いたい領域の大きさに基づいて選択されます。
- 126が最大ですが、メモリの関係上各数値を80に設定しておきます。
- num_modes:
10は、生成されるバインディングモード(結合構造)の最大数を示しています。AutoDock Vinaは、複数の異なるリガンドの結合構造(モード)を生成することができ、これによりリガンドがどのようにタンパク質と結合する可能性があるかを評価できます。
- energy_range:
4は、最良の結合モード(最も低いエネルギー)と表示される最悪の結合モードとの間の最大エネルギー差を示しています。この値が大きいほど、エネルギー的に不利な結合モードも結果として表示される可能性があります。
この設定ファイルを使用してAutoDock Vinaを実行すると、上記の設定に基づいてドッキング計算が行われます。
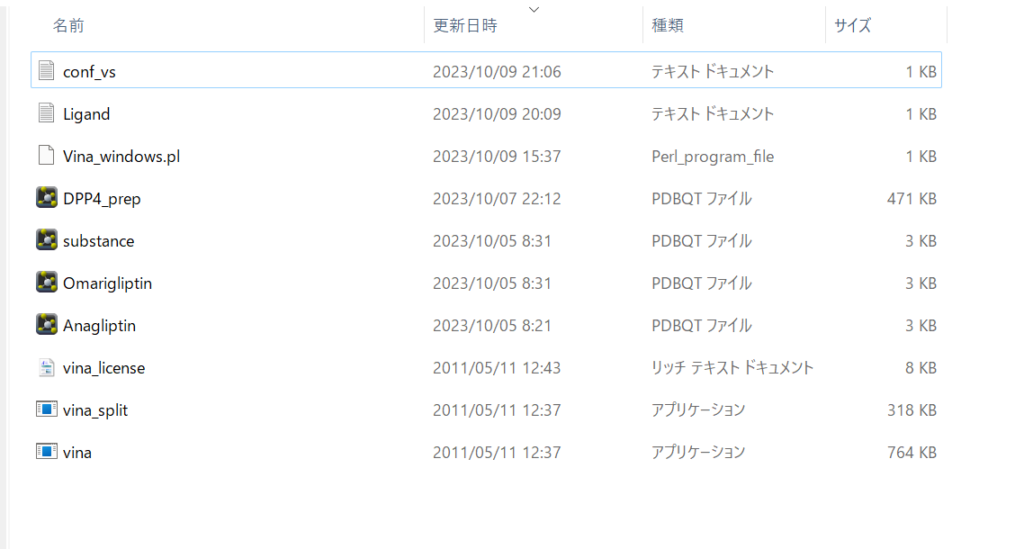
続いて、同様にLigand.txtも作ります。現段階では何も書いてなくて大丈夫です。
現在、作業ディレクトリは以下のようになっていると思います。
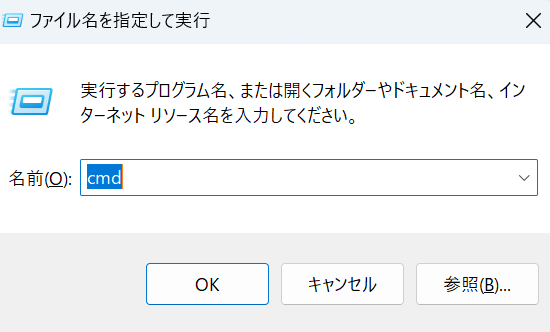
続いて、Windows+rで以下のような画面が開くので、cmdと打ち、コマンドプロンプトを起動してください。
続いて、こちらからperlをダウンロードしてください。dwimperl-5.14.2.1-v7-32bit.exeのファイルです。そして、Perl(Command line)を開いてください。

以下のコードをPerl(command line)で実行してください。
dir /B > Ligand.txt
このコマンドはWindowsのコマンドプロンプトで使用されるもので、現在のディレクトリのファイルとサブディレクトリの名前のみを**Ligand.txt**に出力します。
具体的には:
dir: ディレクトリの内容を表示するコマンド。/B: これはオプションで、名前のみを表示します。通常、**dirコマンドはファイルサイズ、作成日、最終変更日などの詳細も表示しますが、/B**オプションを使用すると名前のみが表示されます。>: 出力のリダイレクト。この記号の後にファイル名を指定すると、コマンドの出力はそのファイルに保存されます。Ligand.txt: 出力が保存されるファイルの名前。
このコマンドを実行すると、**Ligand.txt**という名前のファイルが現在のディレクトリに作成され、その中には現在のディレクトリ内のファイルとサブディレクトリの名前がリストとして保存されます。
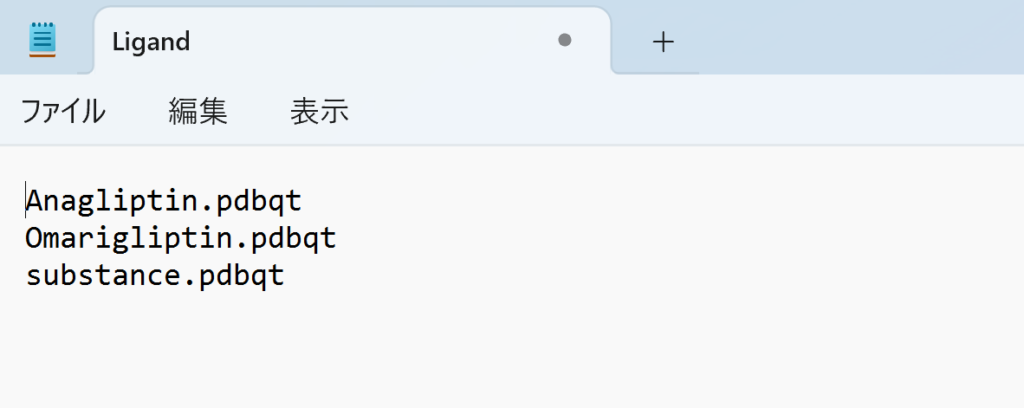
そして、Ligandファイルを開くと、現在のディレクトリの各ファイルの名前が書いてあると思います。リガンドのpdbqtファイル以外は全部消しましょう。今回は三つのリガンドでスクリーニングを行うので、以下の三つのリガンドが書かれています。
Anagliptin.pdbqt
Omarigliptin.pdbqt
substance.pdbqt
さて、これにてin silico screeningの準備は完了です。
In silicoスクリーニング
それでは実際にIn silicoスクリーニングをしていきましょう!
以下のコードを実行してください
perl Vina_windows.pl
Ligandファイルが求められるので、Ligand.txtと入力します。
そして、Enterを押してください。
以下のような画面が出るとスクリーニングが行えています。
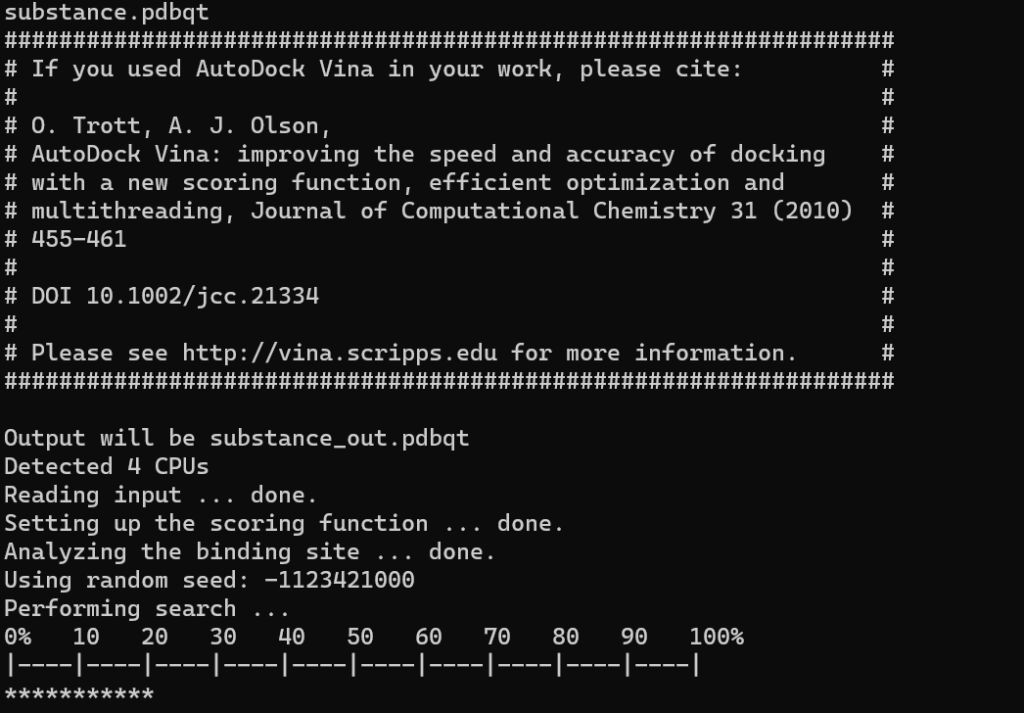
計算が終わると、リガンド名_logファイルとリガンド名_outのファイルが生成しています。
結果
Anagliptin_outとDPP4_prepファイルを開いてみましょう。
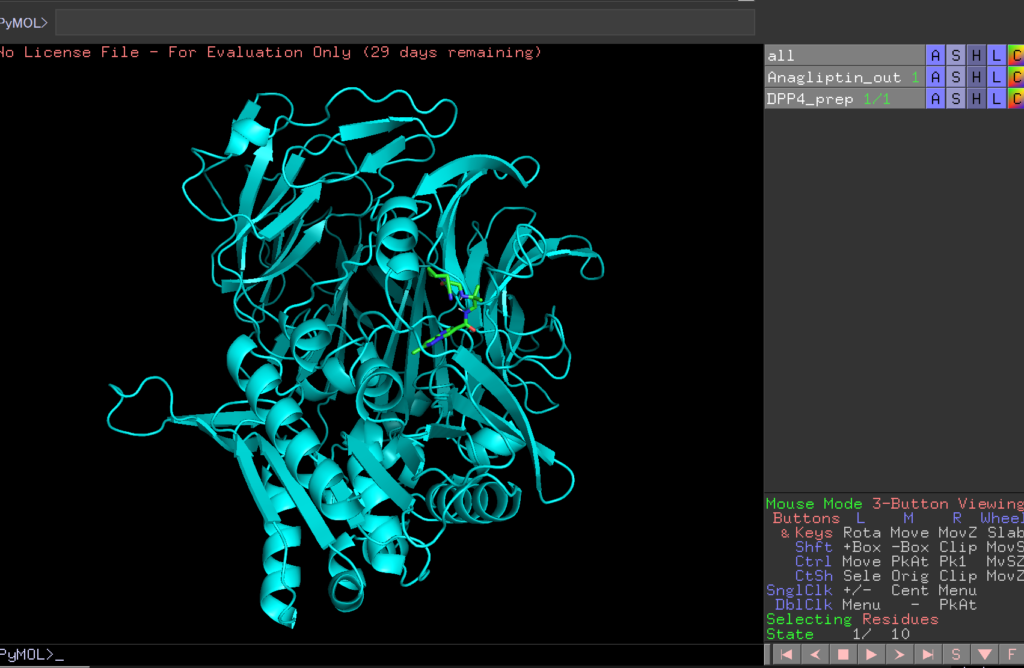
右下にある再生ボタンを押すと、10か所の予測結合サイトが見れます。
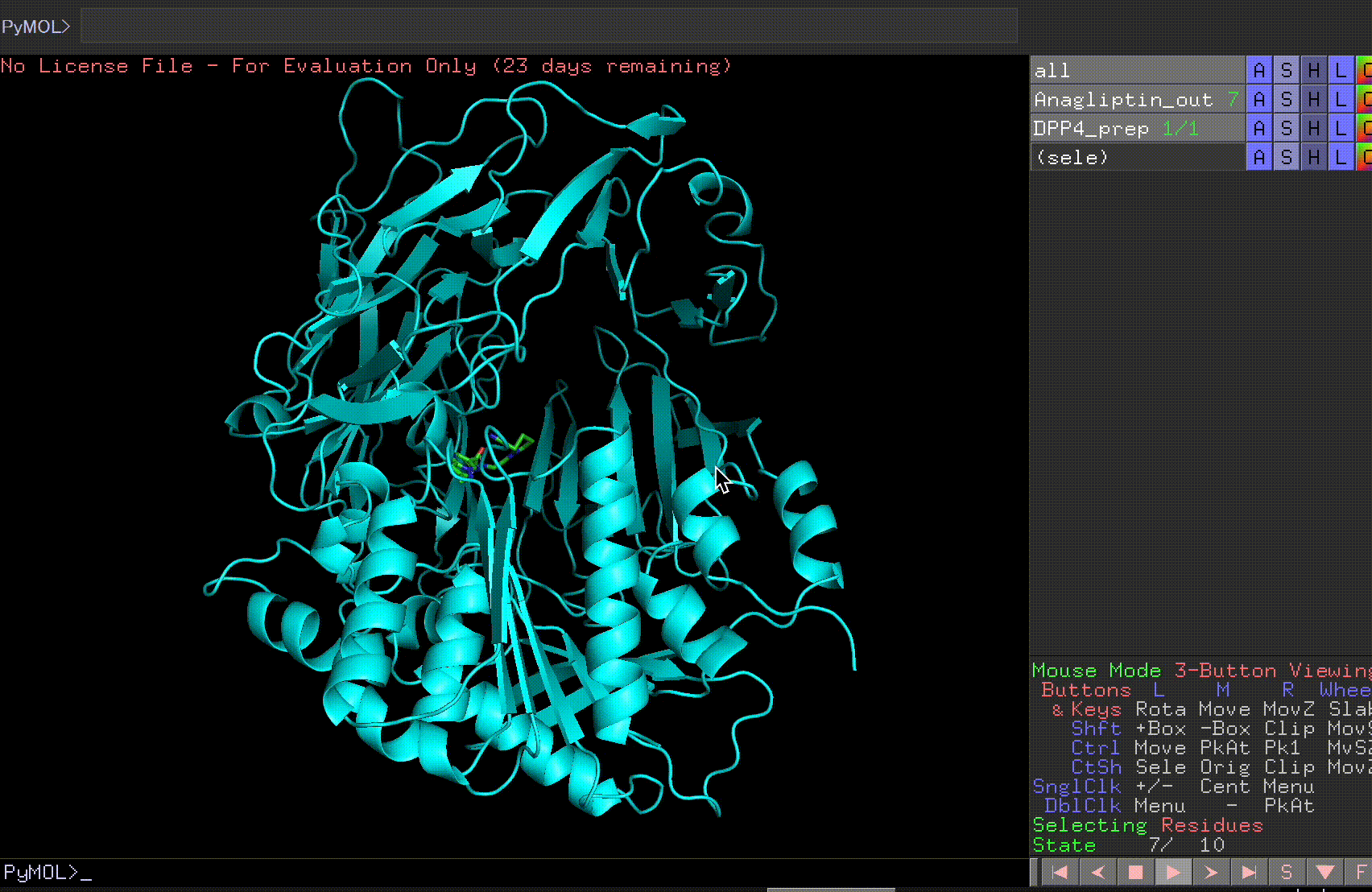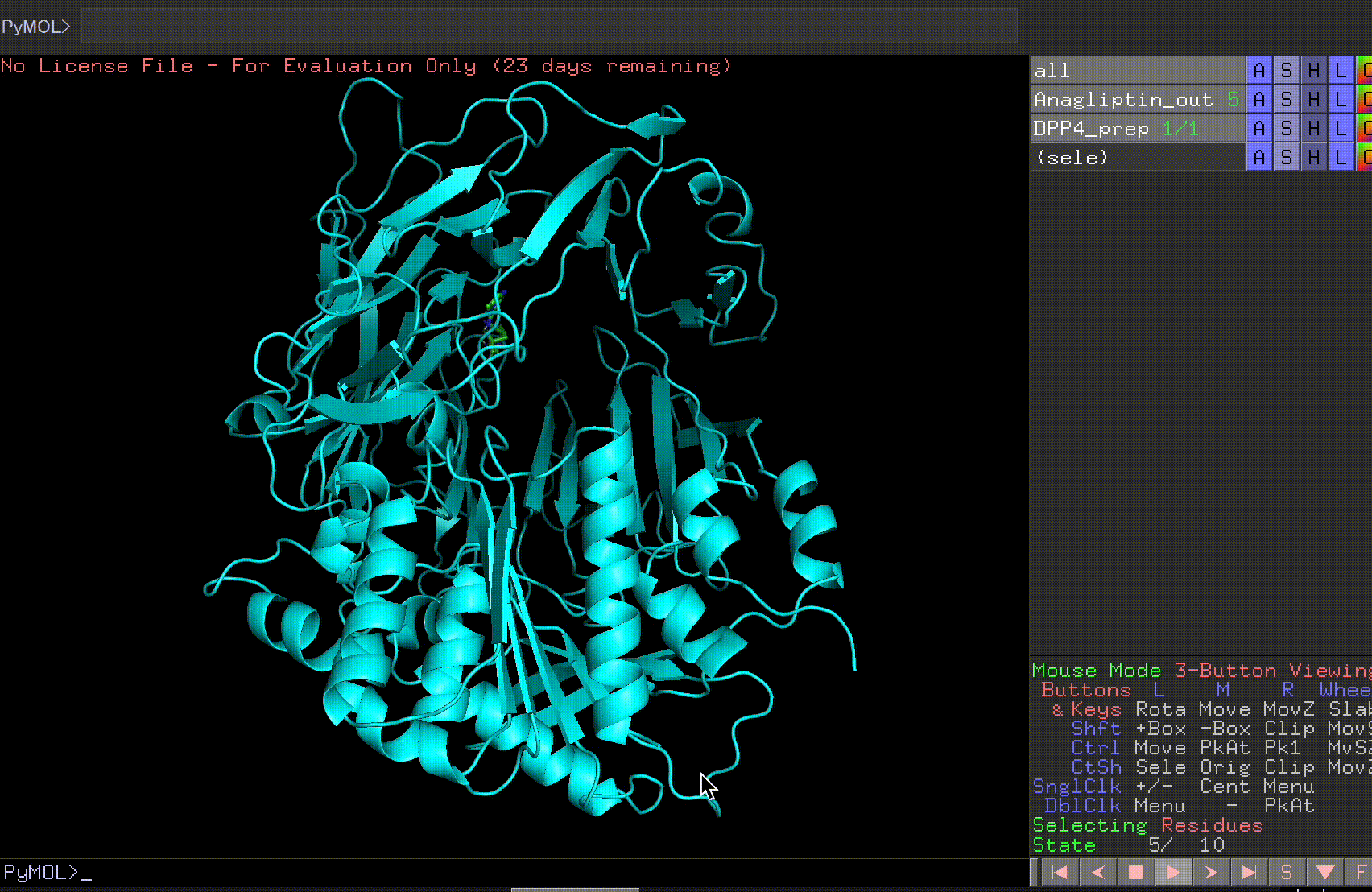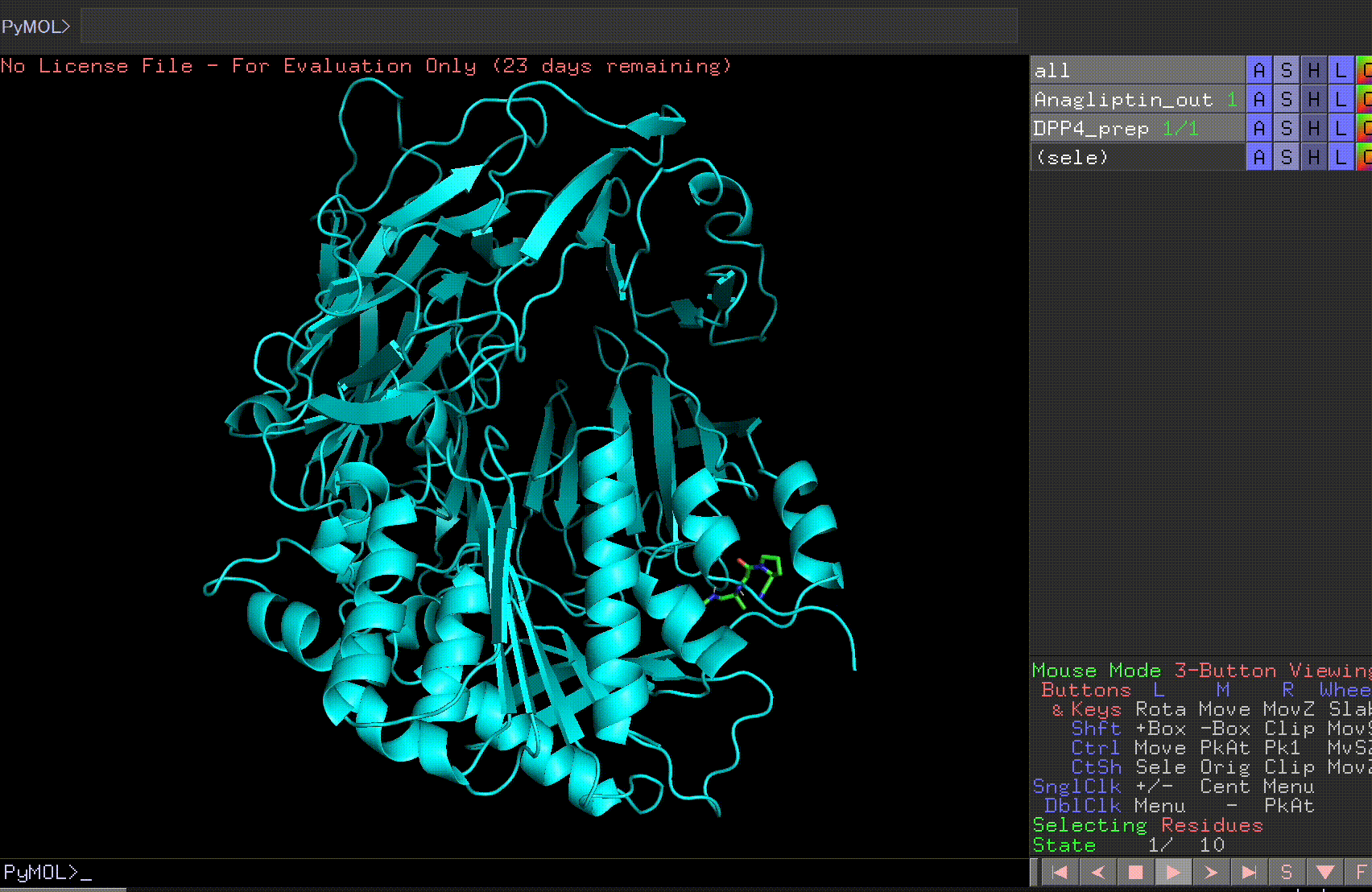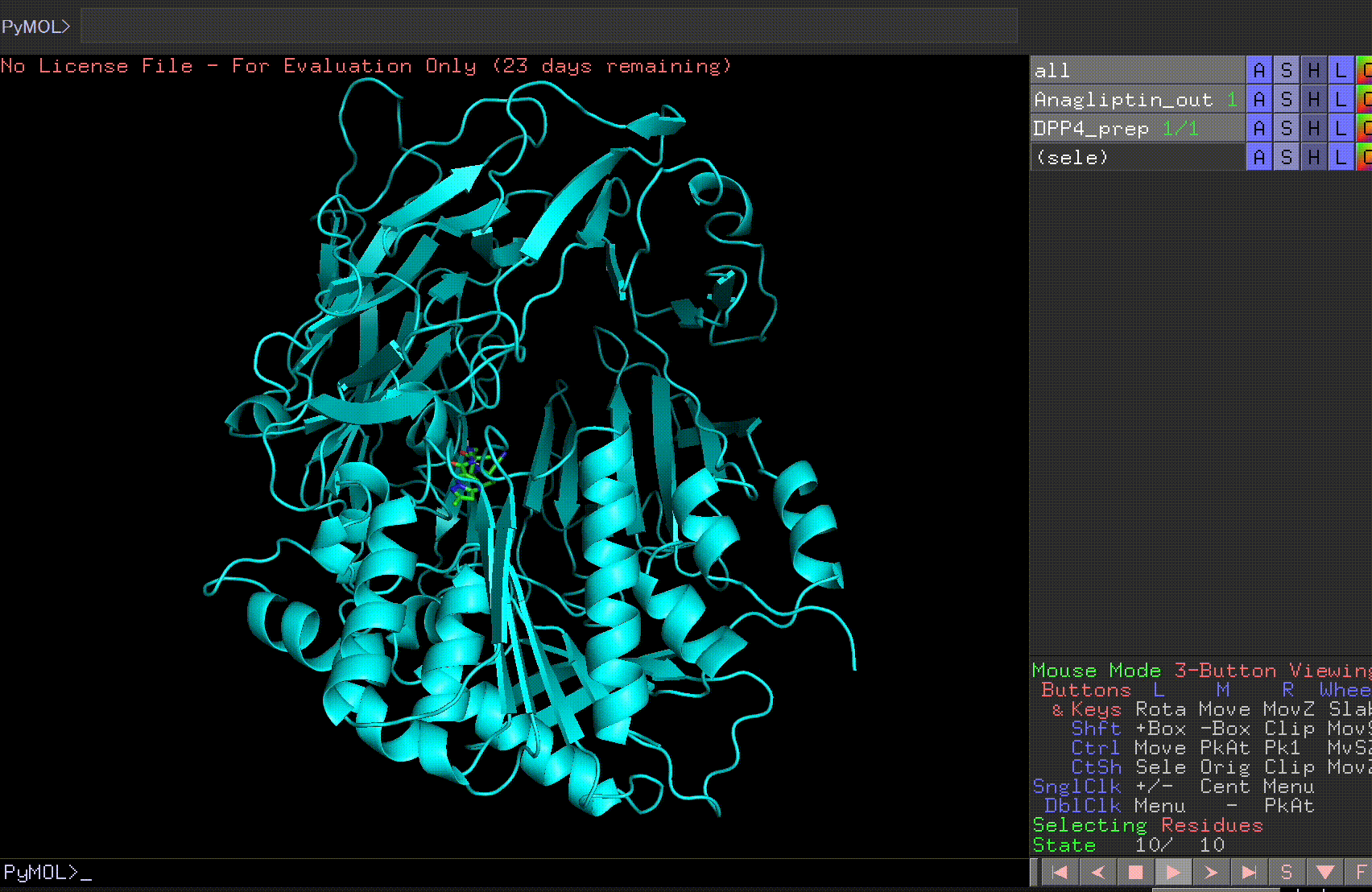
Anagliptin.pdbqt_logで各サイトの結合力を見てみましょう。最も結合力が高いのは-6.9 kcalのものとなりそうです。
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.9 0.000 0.000
2 -6.7 3.102 4.347
3 -6.6 20.983 24.994
4 -6.6 34.775 36.825
5 -6.6 37.303 39.078
6 -6.6 33.968 36.318
7 -6.5 27.023 28.704
8 -6.5 3.644 5.007
9 -6.4 54.846 57.680
10 -6.4 27.367 28.847
このログは、AutoDock Vinaによるリガンドとターゲットタンパク質のドッキング結果を示しています。具体的には、リガンドの複数のドッキング姿勢(mode)の結果を示しており、それぞれの姿勢に関する情報がリストされています。
以下、各列の意味を説明します:
- mode: ドッキングの姿勢を示す番号です。最もエネルギーが低い(最も好ましい)姿勢が1で、次に好ましい姿勢が2、というように続いています。
- affinity (kcal/mol): この姿勢の結合親和性を示すエネルギー値です。この値が低いほど、リガンドとターゲットタンパク質の結合が強いことを示します。
- dist from best mode: この部分は、最も好ましい姿勢(mode 1)との比較に基づく距離を示しています。
- rmsd l.b.: lower bound(最小値)のRMSD (Root Mean Square Deviation) 値。これは、リガンドの姿勢の平均的な偏差を示す値です。
- rmsd u.b.: upper bound(最大値)のRMSD値。
RMSDは、2つの3D構造の間の原子の位置の平均的な偏差を示す値で、小さいほど2つの構造が似ていることを示します。RMSDが大きい場合、それは2つの構造が大きく異なることを示しています。
このログを解釈すると、最も好ましい結合姿勢(mode 1)は、結合親和性が-6.9 kcal/molであり、その後の姿勢はそれと若干異なる形や位置でのドッキング結果を示しています。2番目や8番目の姿勢は1番目の姿勢と比較して、それほど離れていないことを示していますが、3番目の姿勢以降は1番目の姿勢と大きく異なる可能性が高いです。
さて、各リガンドの結合サイトを見てみましょう。
さらにOmarigliptin_outとsubstance_outをpymolで開いてみてください。
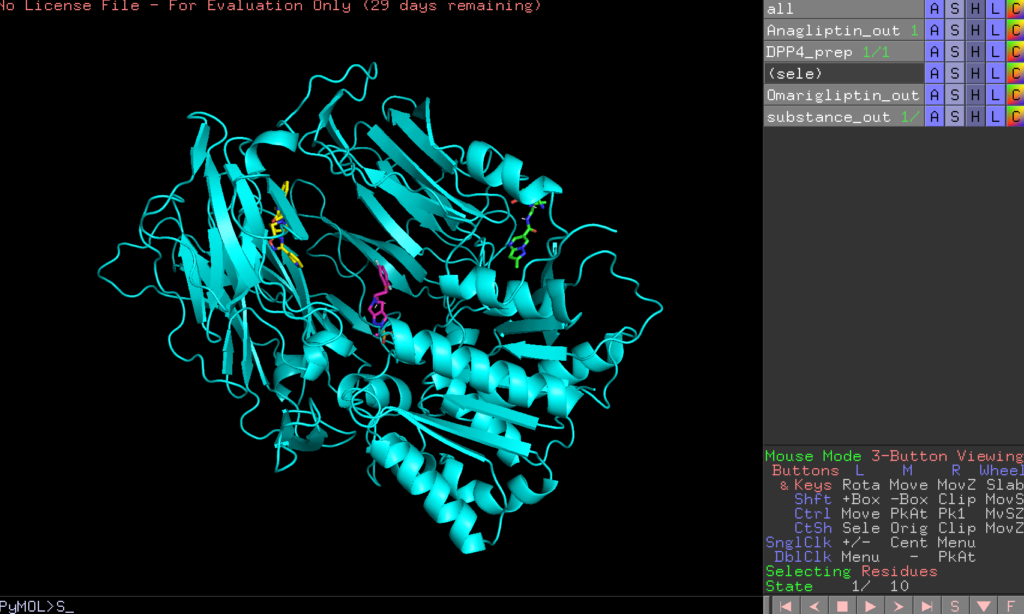
以下では緑がAnagliptin_out、赤がOmarigliptin_out、黄色がsubstance_outです。
各結合力は以下の通りになりました。
Anagliptin_out
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.9 0.000 0.000
2 -6.7 3.102 4.347
3 -6.6 20.983 24.994
4 -6.6 34.775 36.825
5 -6.6 37.303 39.078
6 -6.6 33.968 36.318
7 -6.5 27.023 28.704
8 -6.5 3.644 5.007
9 -6.4 54.846 57.680
10 -6.4 27.367 28.847
Omarigliptin_out
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -8.2 0.000 0.000
2 -7.5 4.813 9.825
3 -7.0 1.396 2.552
4 -7.0 22.600 25.155
5 -6.9 26.790 28.270
6 -6.8 24.370 25.515
7 -6.8 5.341 8.885
8 -6.7 35.981 37.517
9 -6.7 23.764 25.377
10 -6.7 20.055 22.510
substance_out
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -8.1 0.000 0.000
2 -7.9 25.160 28.365
3 -7.9 35.292 37.012
4 -7.9 29.785 33.301
5 -7.8 38.196 39.991
6 -7.8 26.114 28.770
7 -7.7 49.792 53.366
8 -7.7 23.544 28.206
9 -7.7 24.302 27.489
10 -7.7 3.062 6.948
以下に、このデータの解釈を示します。
- 結合親和性: すべてのリガンドの最も好ましい結合姿勢の結合親和性(affinity)を比較すると、Omarigliptinが最も高い親和性を持ち、次にsubstance、最後にAnagliptinとなります。この情報は、リガンドがターゲットタンパク質とどれだけ強く結合するかを示しています。
- ドッキング姿勢の多様性: RMSDの値は、最も好ましい姿勢からの他の姿勢の偏差を示しています。RMSDが低い値を持つ姿勢は、最も好ましい姿勢と構造的に類似していることを示しています。一方、高いRMSD値は、異なる結合モードや異なる結合位置を示しています。
- 結果の信頼性: RMSDの範囲(l.b.とu.b.の間)が狭い場合、その姿勢の結果は比較的信頼性が高いと考えられます。逆に、RMSDの範囲が広い場合、その姿勢の正確さには不確実性があると考えられます。
具体的には、Anagliptinの場合、最も好ましい姿勢から2番目と8番目の姿勢が比較的近いですが、他の姿勢は大きく異なる可能性があります。Omarigliptinでは、1番目から3番目の姿勢が類似しており、substanceでは、1番目と10番目の姿勢が比較的近いです。
既知の薬剤Anagliptinよりも、ランダムに選ばれたsubstanceが高い結合親和性を示すのは驚きの結果です。しかし、substanceの最も高い結合力を持つ1のmodeは他のmodeと比べて数が少ないようです。これは、substanceがターゲットに結合する際、最も高い結合力を示す姿勢よりも、やや低い結合力を持つ他の姿勢をとることが多いことを意味します。この観点から考えると、Omarigliptinの方が全体的に安定して高い結合力を持っていると考えられます。
最後に
いかがでしたでしょうか?以前に紹介したPyRxは今回のAutodock vinaによるスクリーニングをよりやりやすくしたもののになります。今回の操作は少し難しかったので、慣れない方はこちらで行うもありだと思います!
このようにコンピュータ上で簡単にin silicoスクリーニングを行うことができます。ぜひ皆さんも試してみて、創薬を体験してみてください!
参考文献
ドッキングシミュレーションのやり方【AutoDock vina】 | 計算化学ポータルサイト | 計算化学.com
AutoDock Vina: Molecular docking program — Autodock Vina 1.2.0 documentation

