
This article is about SwissDock, a low-molecular-weight protein docking software. Understanding this content will enable you to predict complexes of desired compounds and proteins. I encourage everyone to give it a try!
Windows 11 Home, UCSF Chimera 1.17.3
Before proceeding with this article, please download UCSF Chimera.
What is Molecular Docking?
Molecular docking is a critical method in drug development. It involves simulating the interaction between large molecules, such as proteins, and small molecules (potential drugs) on a computer. Molecular docking predicts how a drug binds to its target protein and assesses the strength and characteristics of the binding. This helps understand the effects and side effects of new drug candidates, streamlining screening in the early stages of drug development. Molecular docking is useful for identifying promising drugs from a vast library of potential compounds.
About Swiss-Dock
Swiss-Dock is a free, web-based tool for performing molecular docking. It focuses particularly on modeling protein-ligand interactions. Users upload the 3D structures of the target protein and ligand (potential drug molecule), and Swiss-Dock predicts the optimal binding modes between these molecules. The results are provided as visual representations, helping evaluate where and how the ligand binds to the protein, the shape of the binding, and the strength of the bond. Swiss-Dock is widely used by researchers and students for its ease of use and accessibility.
Molecular Docking with Swiss-Dock
Preparing the Protein
First, we’ll prepare the protein.
We will use the target protein MPro of the novel coronavirus (PDB: 2OPQ) mentioned in this blog.
Please download the PDB Format.
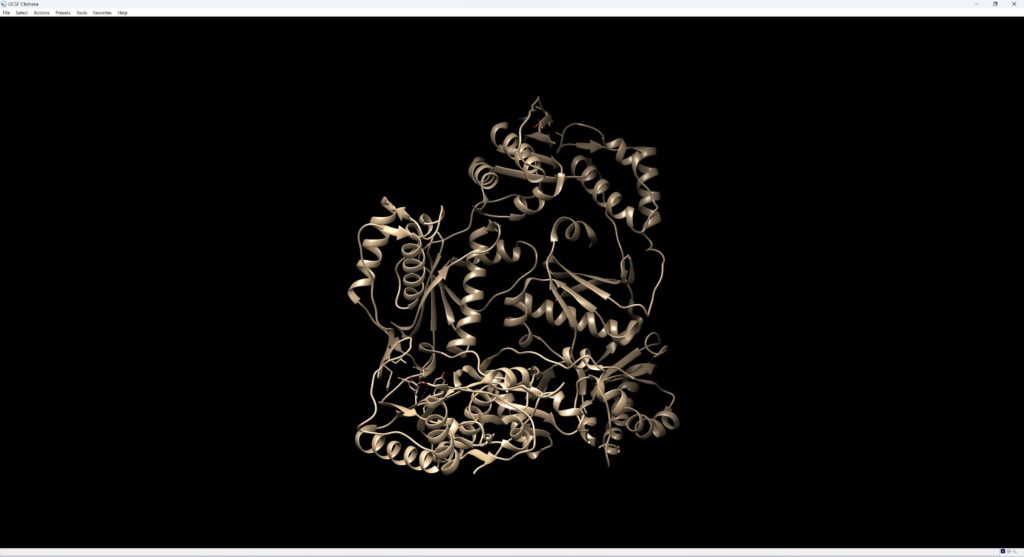
After downloading, open the file in UCSF Chimera. It should look like the image below.
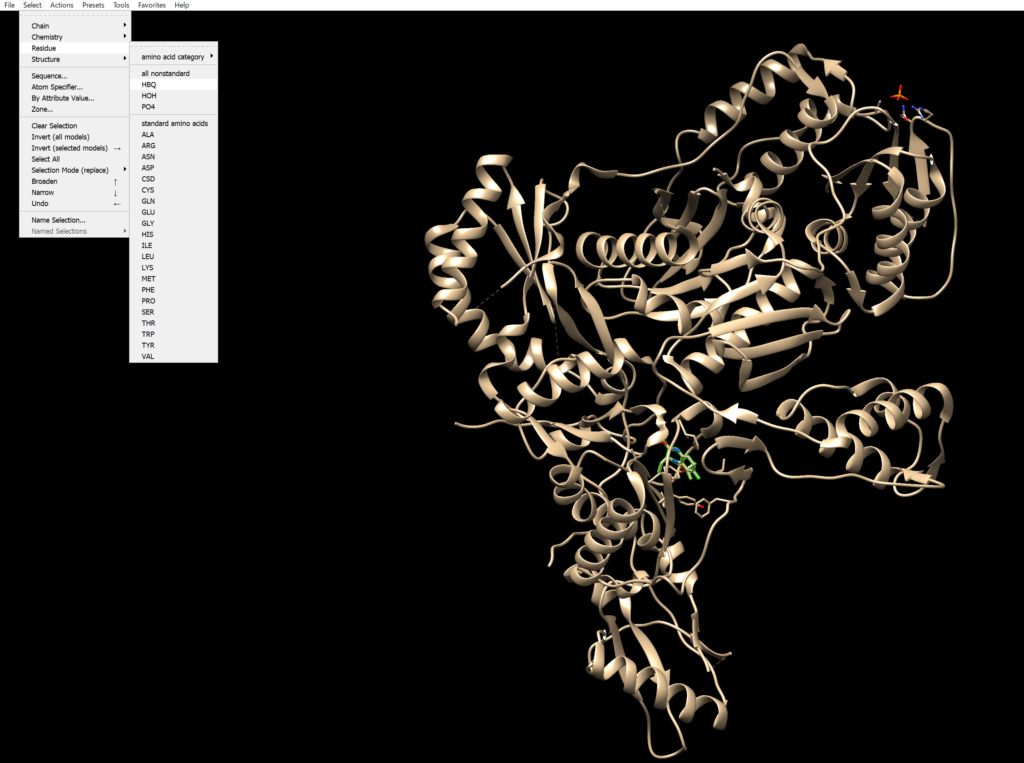
This protein contains non-amino acid elements, which we will remove.
Select HBQ by navigating to Select → Residue → HBQ, and delete it using Action → Delete. Similarly, remove HOH and PO4.
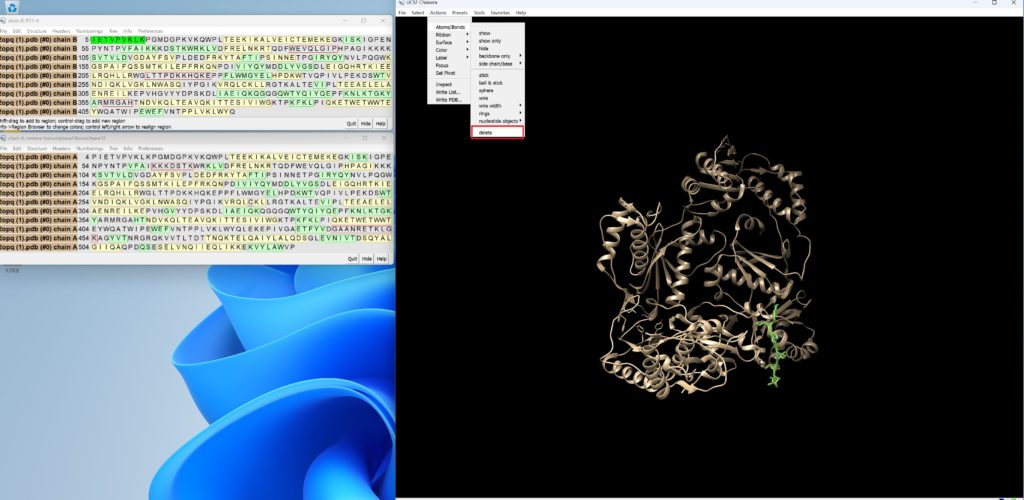
Since the protein is slightly larger than the maximum size for molecules set in SwissDock, we will trim the terminal amino acids slightly.
Open the sequence by going to Tool → Sequence → Sequence, select up to IETVPVKLKPGMDG, and delete it.
Then, save this PDB file using File → Save PDB.
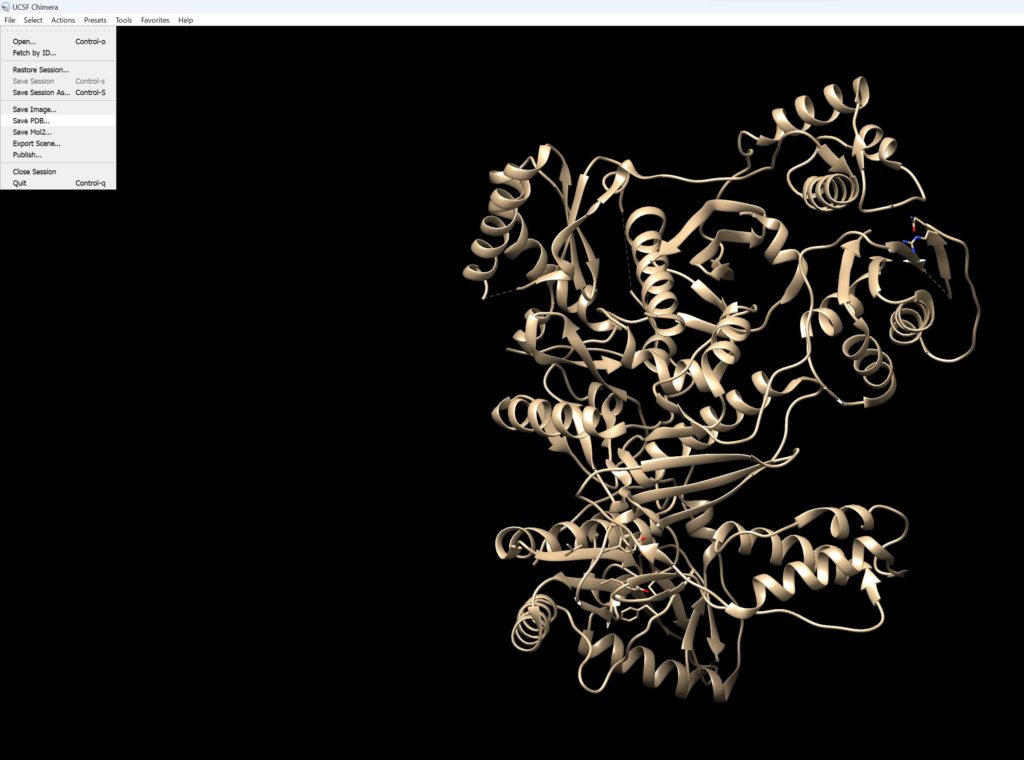
This concludes the protein preparation.
Preparing the Ligand
Next, let’s prepare the ligand. The compound we obtained from previous screening is Tolnaftate.
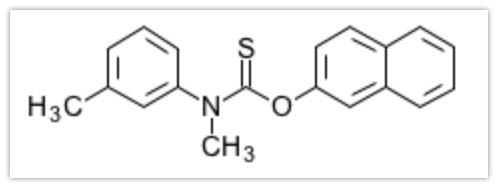
Now, let’s create a file for Swiss-Dock.
Go to the site draw molecule online, draw Tolnaftate, and click Click to Convert.
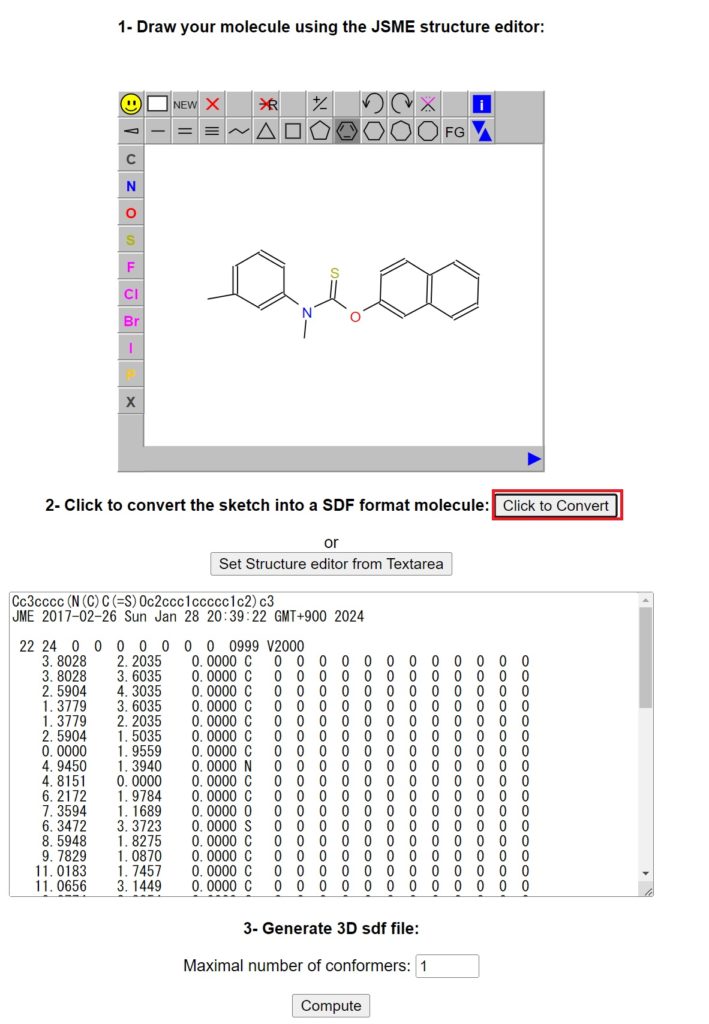
Then, press Compute.
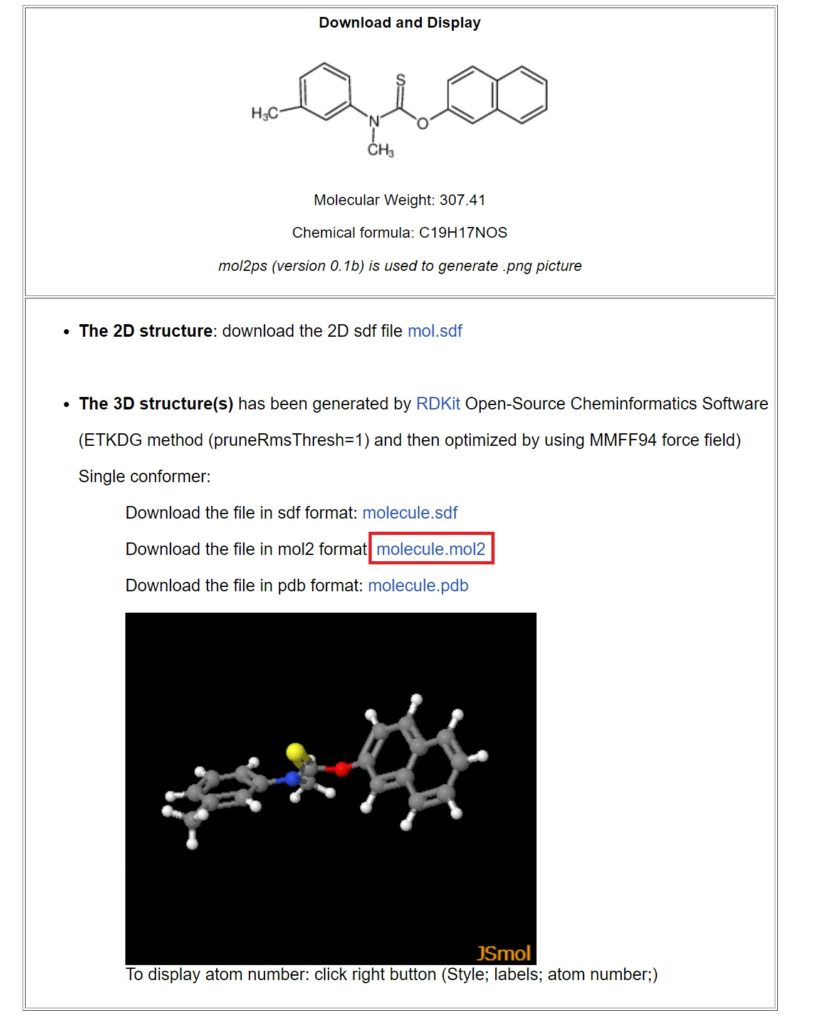
Clicking on molecule.mol2 will download the mol file. Save it as Tolnaftate.mol2.
This concludes the ligand setup.
Molecular Docking with Swiss-Dock
Let’s proceed with molecular docking using Swiss-Dock. Accessing the site should bring you to the screen shown below.
Go to the Submit Docking tab, upload the prepared files, and enter the Job Name and E-mail address.
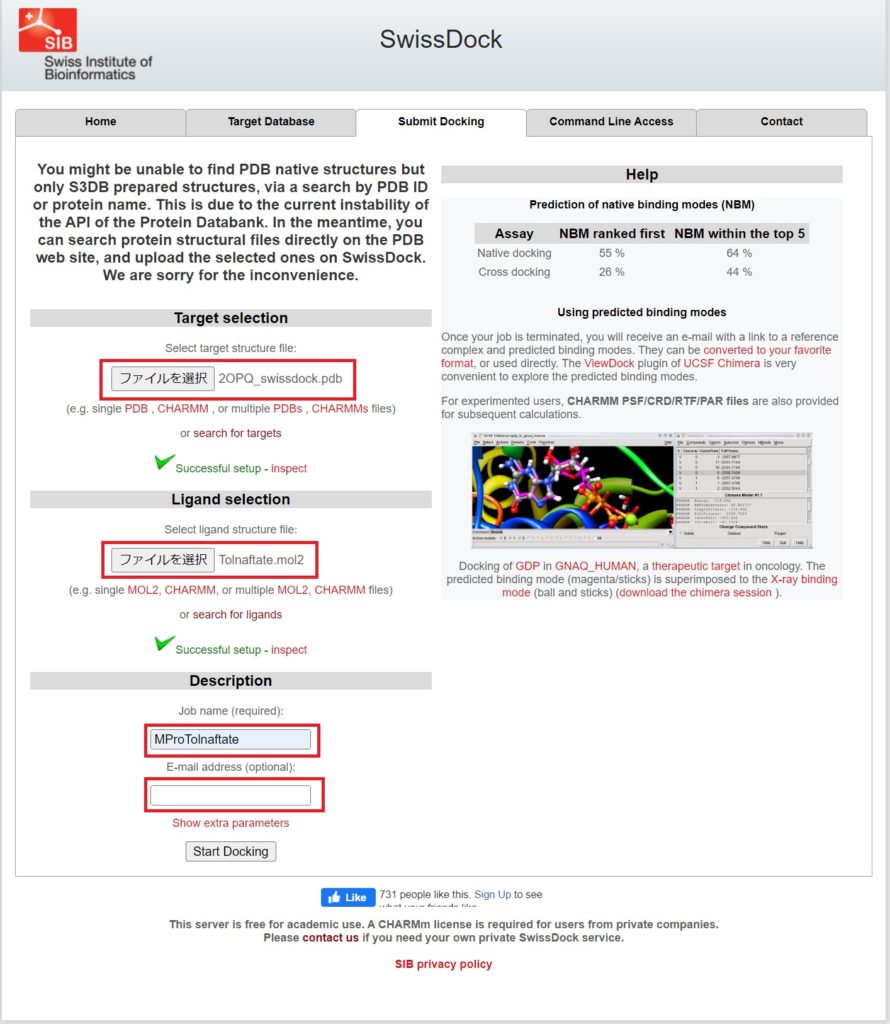
Press Start Docking to begin the molecular docking process.
Once finished, you will receive an email. Open it to see the results.
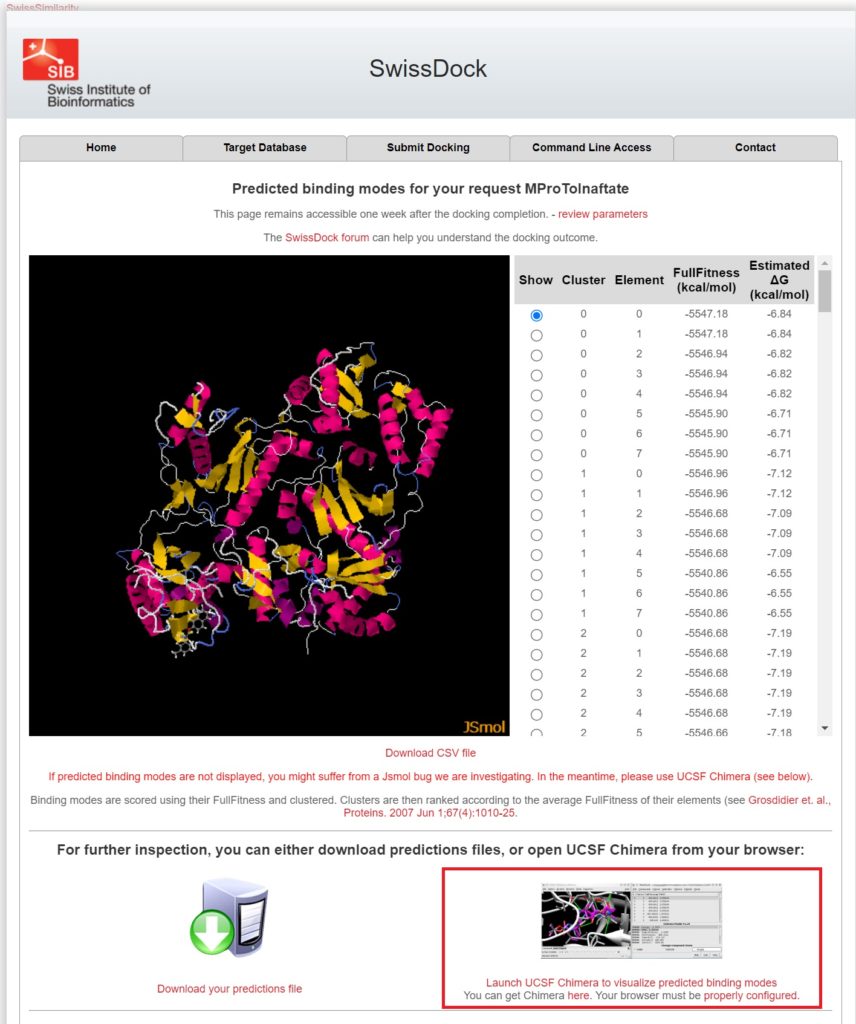
The binding modes are scored and clustered by two values: FullFitness (kcal/mol) and Estimated ΔG (kcal/mol). These values indicate the quality of the docking results, with lower values meaning a stronger predicted binding. The list displays multiple clusters and their energy scores.
For further analysis, click Launch UCSF Chimera to visualize predicted binding modes at the bottom right and open it in UCSF Chimera.
Analysis with UCSF Chimera
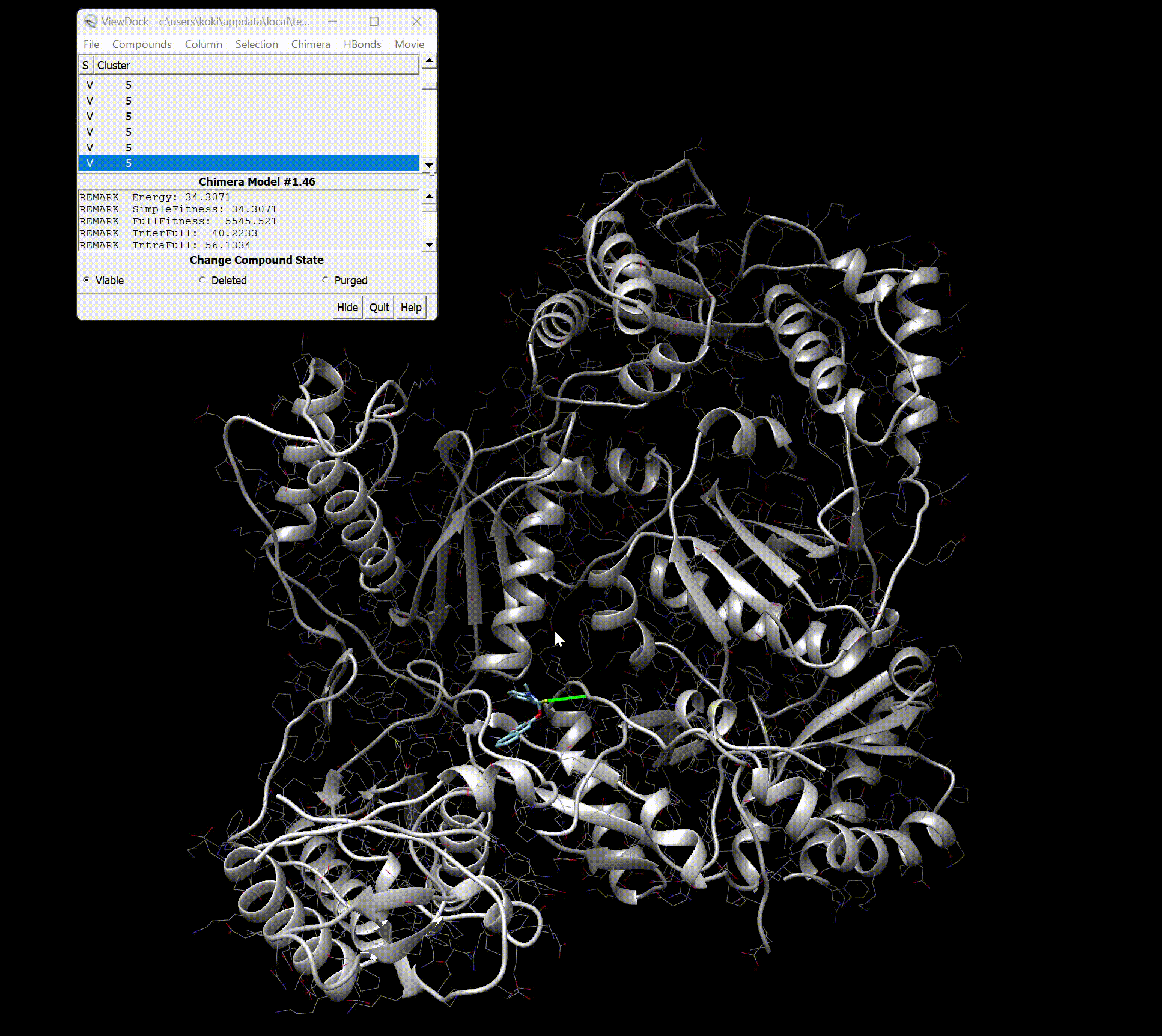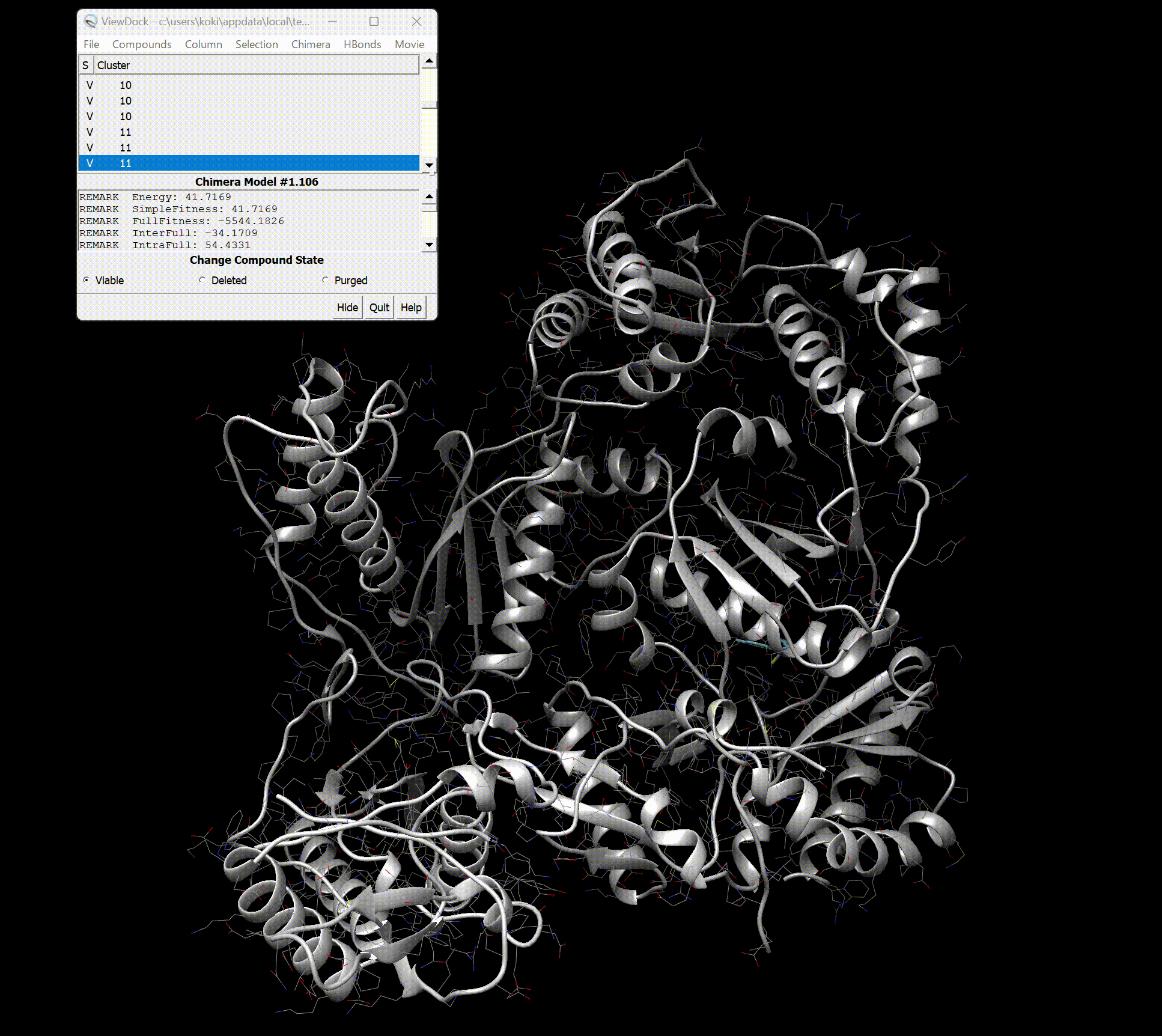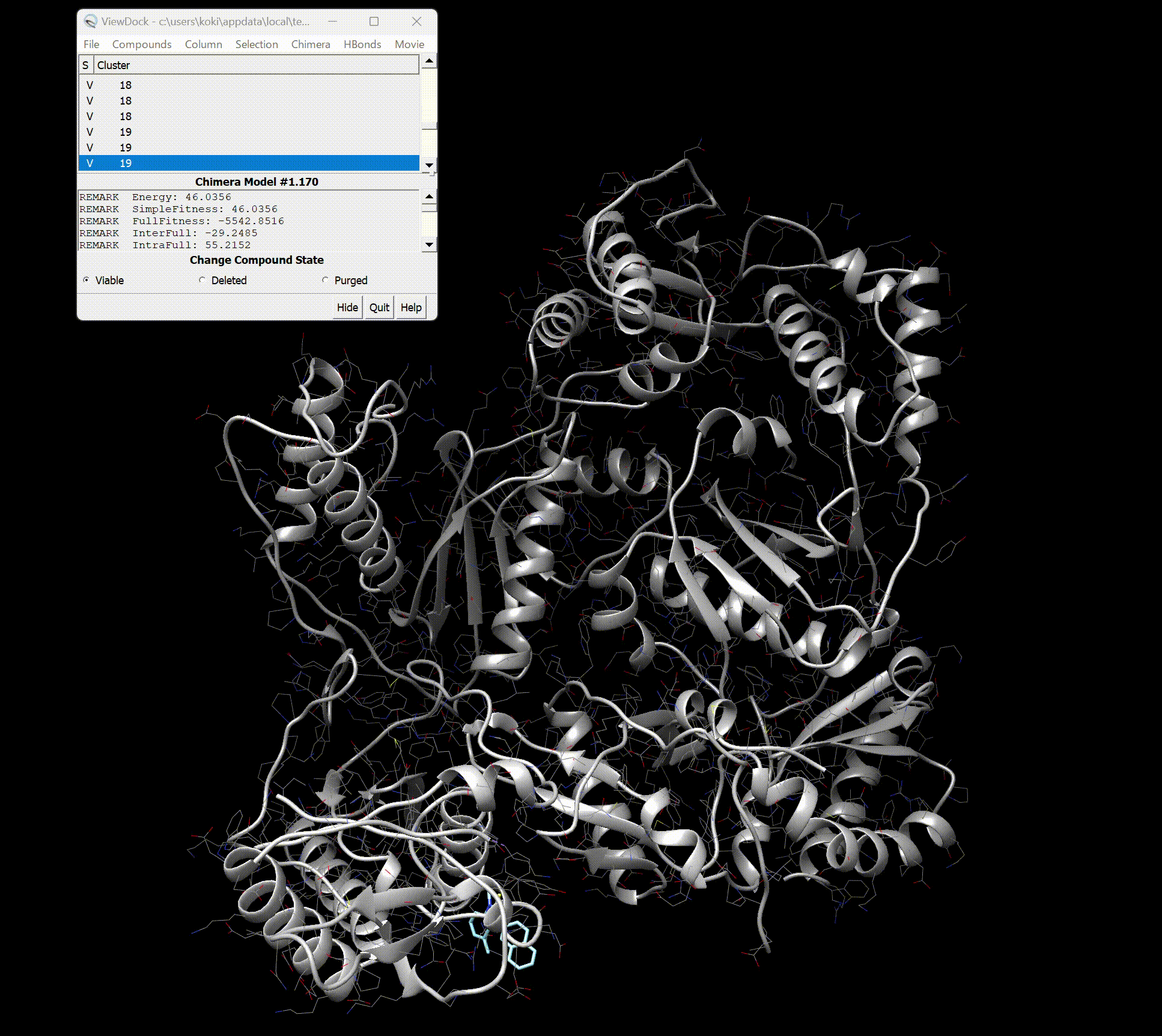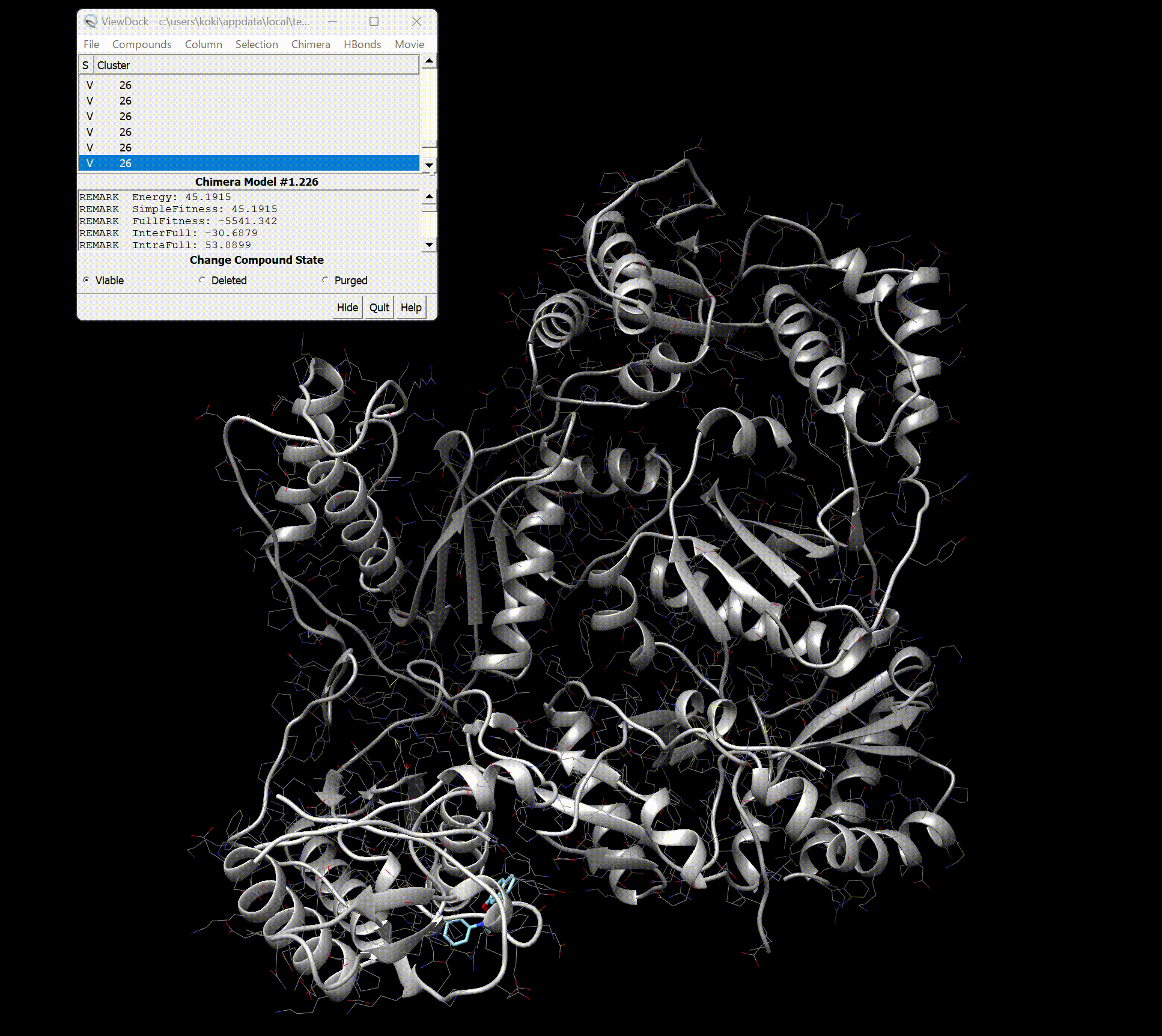
Upon opening, you should see the screen as shown below.
You can see detailed information about each binding site.
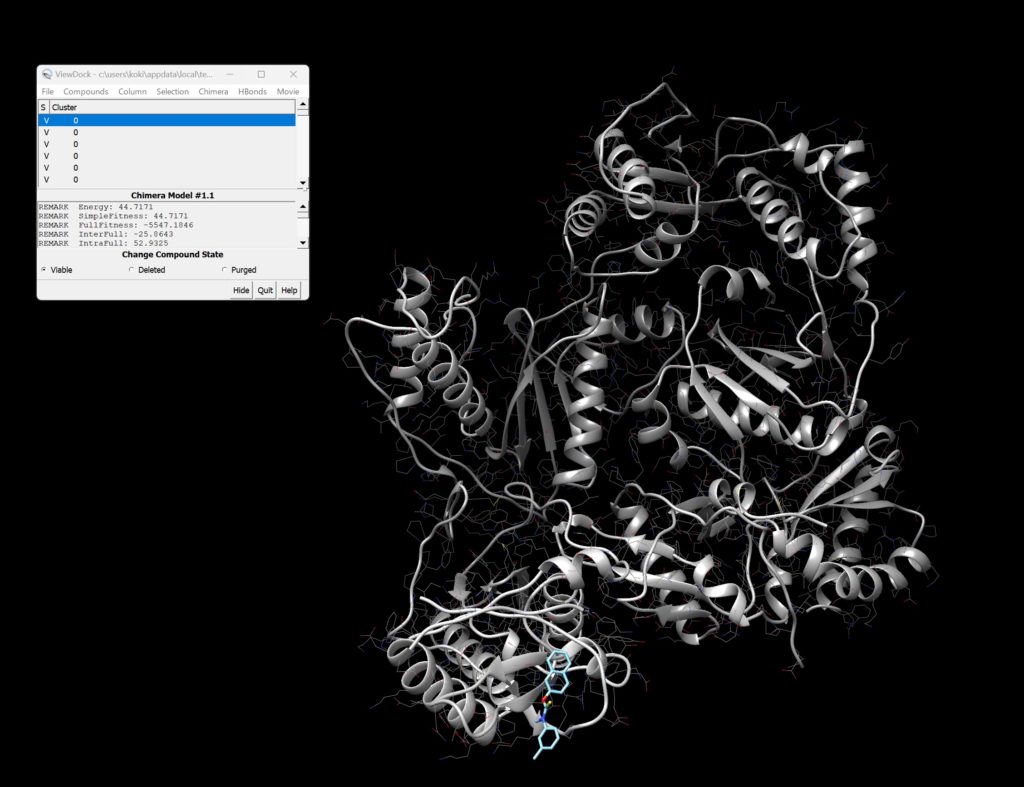
Furthermore, by pressing Movie → Play in the top right, you can view a movie of the binding styles.
In Conclusion
How was it? We have introduced various screening technologies, such as Auto Dock, PyRx, and Machine Learning! Try molecular docking with the compounds you’ve found using these methods for more detailed docking experiments!
References
バイオインフォマティクス: Swissdock タンパク質リガンドのドッキング |バイオコード株式会社
Lesson 12: Protein docking with SwissDock and UCSF-Chimera
SwissDock, a protein-small molecule docking web service based on EADock DSS.