
Many might be interested in trying out single cell RNA-seq (scRNA-seq), but the lack of access to sequencing equipment and unfamiliarity with the analysis methods can be major hurdles.
This article explains how to find scRNA-seq data from public databases.
By reading this article, one can find scRNA-seq data related to their research from public sources. Although the introduction might seem lengthy, it is crucial to understand all the premises thoroughly.
Please note that the scRNA-seq analysis discussed in this article refers to data analysis using scRNA-seq data, and not to sample preparation.
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB

Beginner-Friendly Technical Book on Single Cell RNA-seq Analysis Using Public Data Available for Sale
¥3,600 → ¥1,800 50%OFF!!
Easy-to-Understand Explanation for Programming Beginners!
Start Single Cell RNA-seq Analysis with R and Seurat!
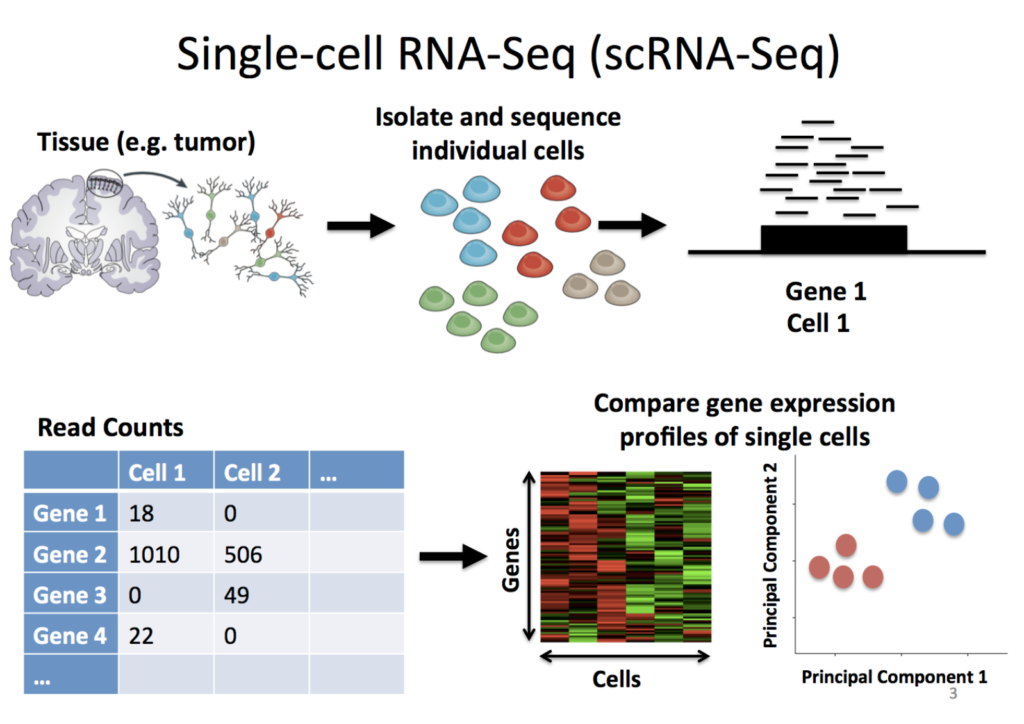
What is scRNA-seq?
Single-cell RNA sequencing (scRNA-seq) is a method used to study gene expression patterns within individual cells. This enables the analysis of gene expression in individual cells within a population of cells. It is commonly used for identifying cells, understanding cell life cycles, and studying cell development.
Stephanie Hicks
Welcome to the World of Single-Cell RNA-Sequencing
https://speakerdeck.com/stephaniehicks/welcome-to-the-world-of-single-cell-rna-sequencing?slide=3
When performing scRNA-seq analysis, which language should you choose: R or Python?
When deciding between R and Python for single-cell RNA-seq analysis, both languages offer packages suitable for the task. The choice between them depends on their respective strengths and weaknesses, as well as your needs and experience.
- Python: Examples include Scanpy and CellRanger.
- Scanpy is suitable for general single-cell RNA-seq analysis.
- CellRanger is tailored for analyses specific to 10x Genomics formats.
- R: Examples include Seurat and Monocle.
- Seurat is suitable for general single-cell RNA-seq analysis.
- Monocle is better for handling time-series data.
Essentially, either language can be a good choice, and it’s advisable to select the one you are more familiar with. If using Python, Scanpy is a solid choice for library analysis, and for R, Seurat is recommended.
About scRNA-seq Databases
scRNA-seq data is registered in several public databases, allowing for the search and access of scRNA-seq data.
- NCBI GEO: Operated by the NCBI, it provides not only bulk RNA-seq data but also scRNA-seq data.
- Single Cell Expression Atlas: A scRNA-seq database managed by EMBL-EBI.
- Single Cell Portal(SCP): A scRNA-seq database run by The Broad Institute of MIT and Harvard. As of the end of January 2023, it contains data from 507 research projects and expression data for 29,614,655 cells.
- The Human Cell Atlas (HCA): Contains scRNA-seq data from human-derived cells.
- The Mouse Cell Atlas(MCA): Contains scRNA-seq data from mouse-derived cells.
- SCPortalen: Operated by RIKEN, focusing on scRNA-seq databases for mouse and human cells.
10x Genomics Format
10x Genomics is a company that provides single-cell RNA sequencing technology, which has become widely popular among researchers due to its ability to analyze a large number of cells at a low cost.
The data generated by 10x Genomics is in a proprietary format. To analyze this unique format, 10x Genomics offers a special library called Cell Ranger, although it can also be analyzed using Seurat. For this reason, we will proceed with the analysis using Seurat.
The file structure of data in the 10x Genomics proprietary format is as follows:
$ tree filtered_feature_bc_matrix
filtered_feature_bc_matrix
├── barcodes.tsv.gz
├── features.tsv.gz (or genes.tsv.gz)
└── matrix.mtx.gzIn this article, we will look for such files and conduct a quality check analysis.
Finding scRNA-seq Data
After a lengthy introduction, this guide aims to demonstrate how to find scRNA-seq data from public databases and proceed to quality check analysis using Seurat. It is beneficial to have prior knowledge of RStudio and Seurat before proceeding, as outlined in the article:
【scRNA-seq】 How to Start scRNA-seq Analysis Using Seurat (Part 1)【Seurat】
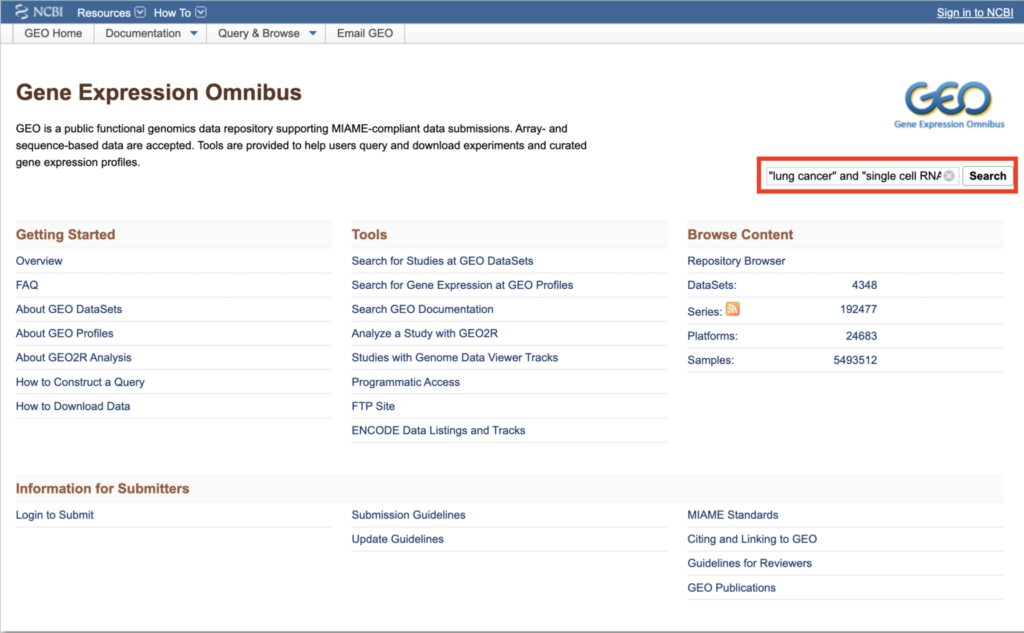
To begin searching for scRNA-seq data, this guide suggests starting with the NCBI GEO database. Please visit the following URL:
https://www.ncbi.nlm.nih.gov/geo/
In the search box located at the top right, enter the search terms as follows:
“[desired search term]” and “single cell RNA-seq”
For this guide, use “lung cancer” and “single cell RNA-seq” as the search terms.
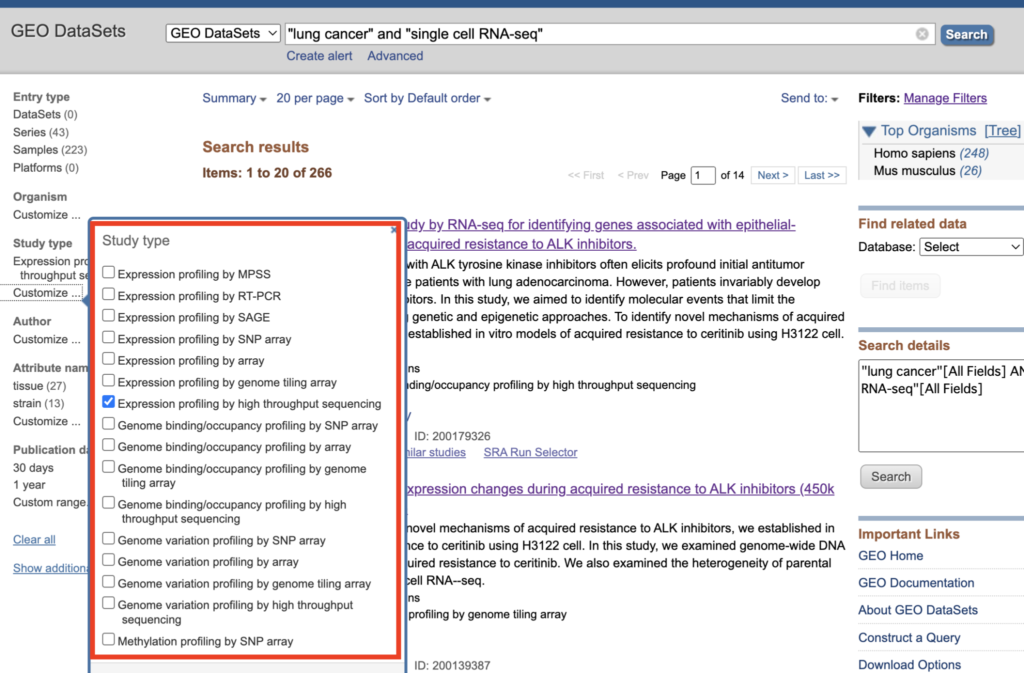
Click on “customize” from the left sidebar and check the box for “Expression profiling by high throughput sequencing”.
After checking, click on “Expression profiling by high throughput sequencing” when it appears.
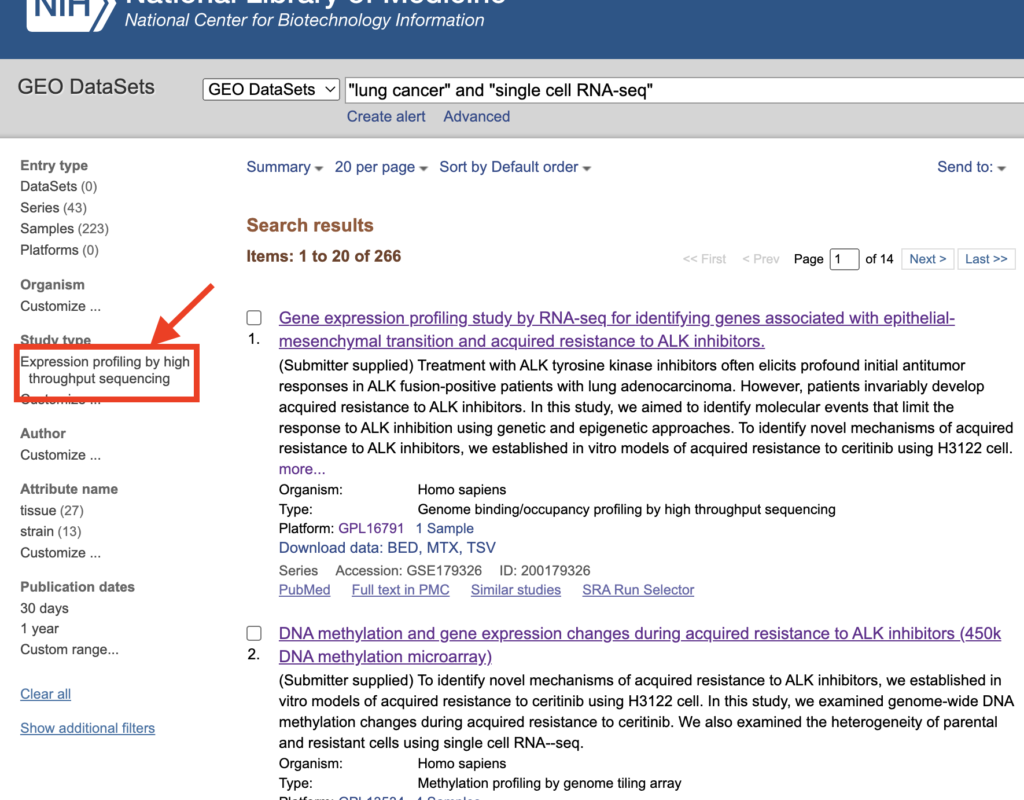
With this preparation complete, the next step involves searching for samples suitable for scRNA-seq analysis. As a trial, consider examining the study at the top, “DNA methylation and gene expression changes during acquired resistance to ALK inhibitors (scRNA-seq).”
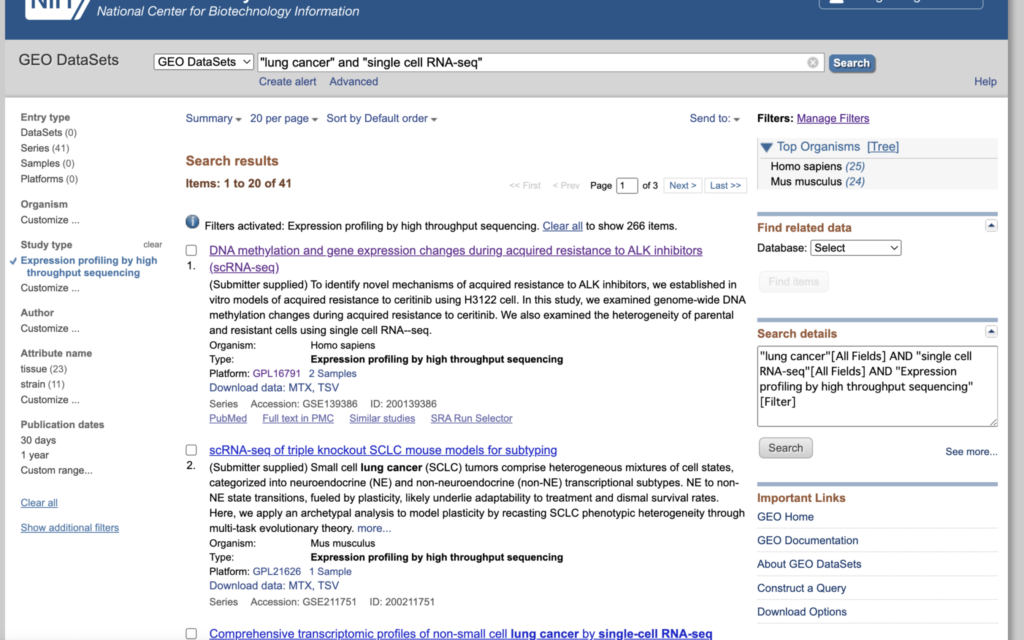
Scrolling down reveals information about the Supplementary file. Clicking on “custom” allows viewing of the file contents. For this sample, the files named barcodes, features, and matrix, which are in the 10X Genomics format, are available and can be used for the analysis.
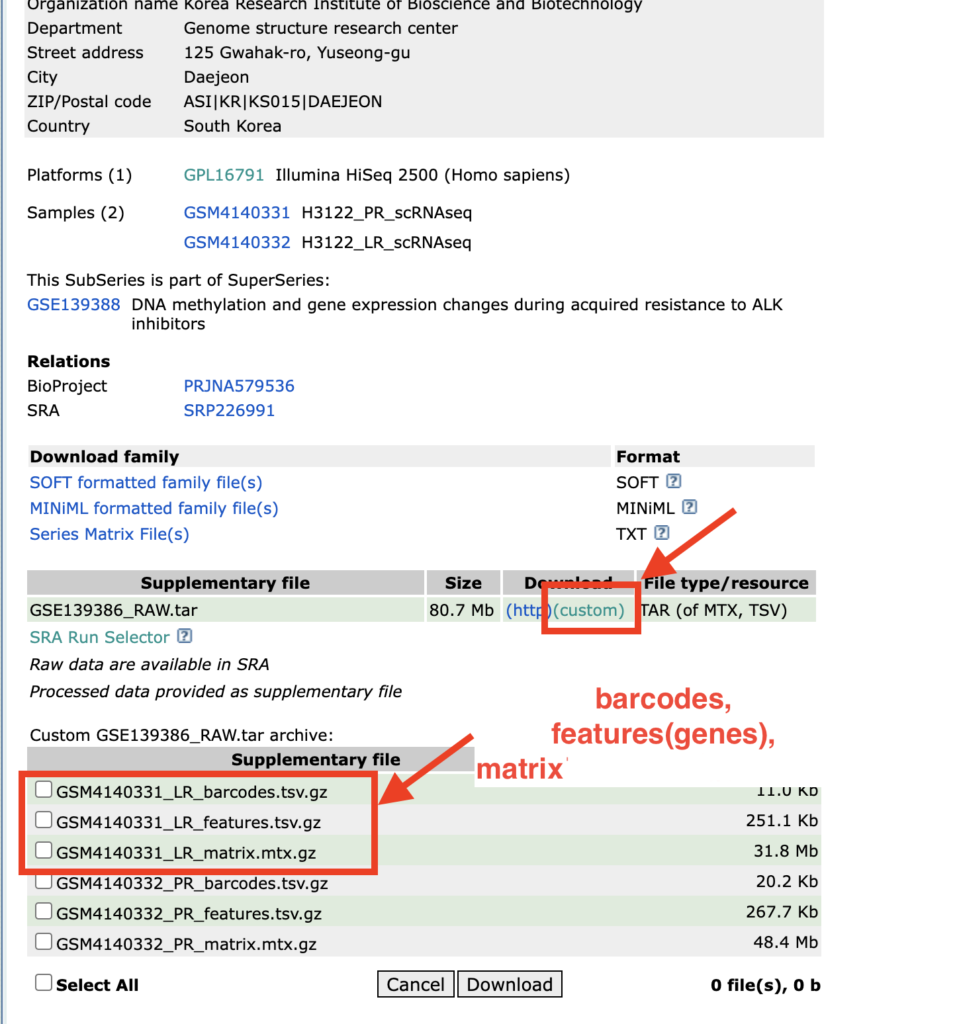
Next, an example of a pattern unsuitable for this analysis will be introduced. Consider looking at “Comprehensive transcriptomic profiles of non-small cell lung cancer by single-cell RNA-seq” found in the search results. Upon examining the contents of the RAW.tar file, it contains txt.gz files. Such a pattern cannot be used for this analysis, so it should be avoided. This will be addressed in a different context at another time.
Now, let’s proceed to analyze “DNA methylation and gene expression changes during acquired resistance to ALK inhibitors (scRNA-seq).” By checking the checkbox next to the file and clicking Download, the files will be downloaded as a tar file. Please bring it to the desktop and unzip it.
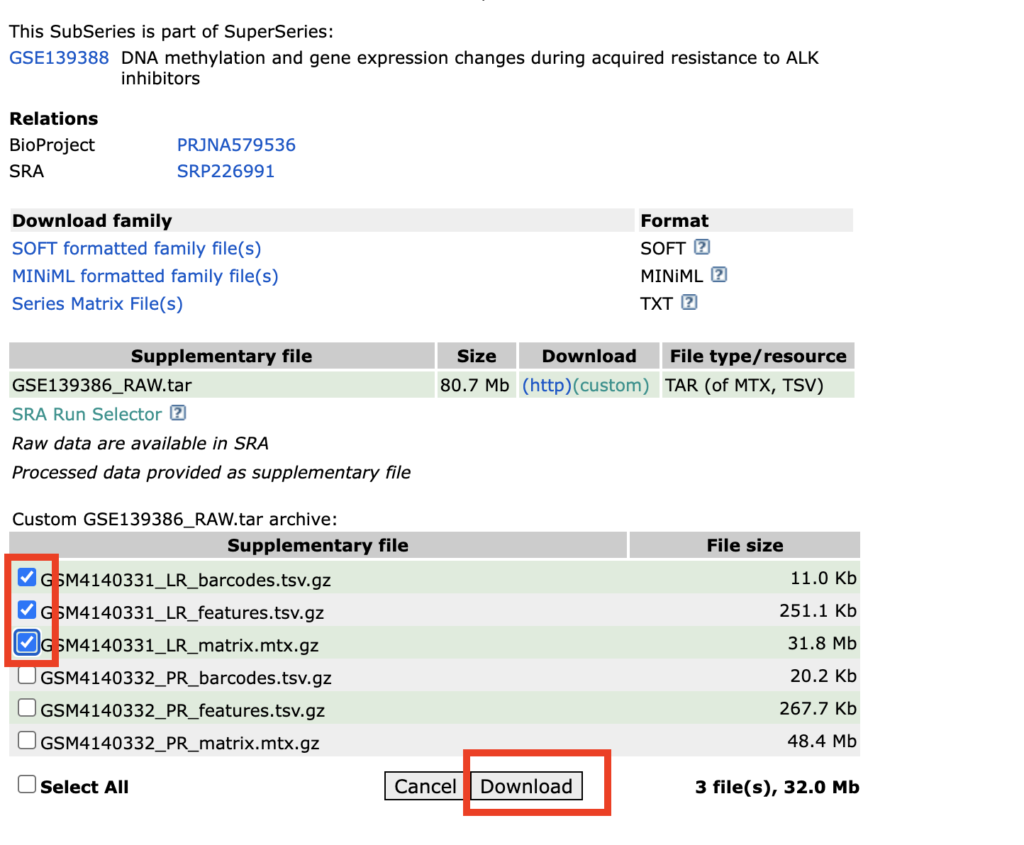
The contents of the downloaded sample will appear as follows:
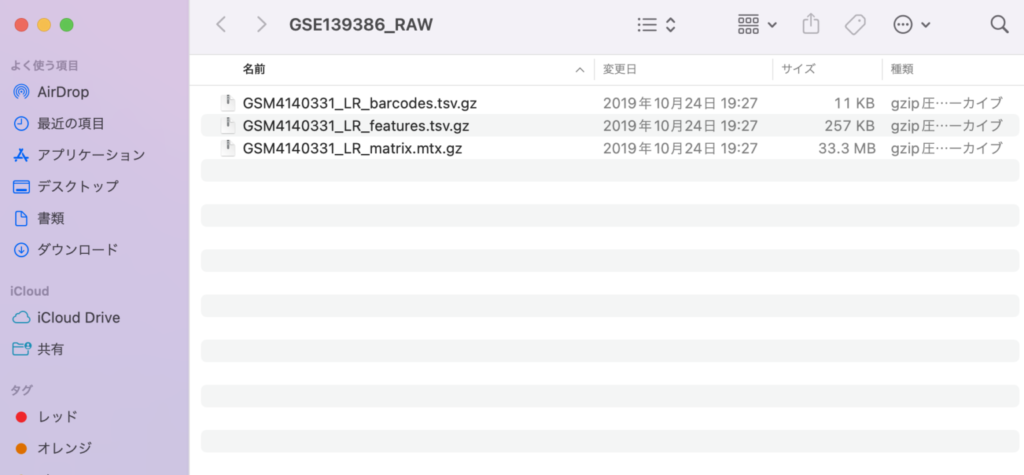
Next, please rename the files to barcodes.tsv.gz, features.tsv.gz, and matrix.mtx.gz, respectively. This step is necessary for loading the files into Seurat. It’s okay to keep the files in gz format; there is no need to unzip them.
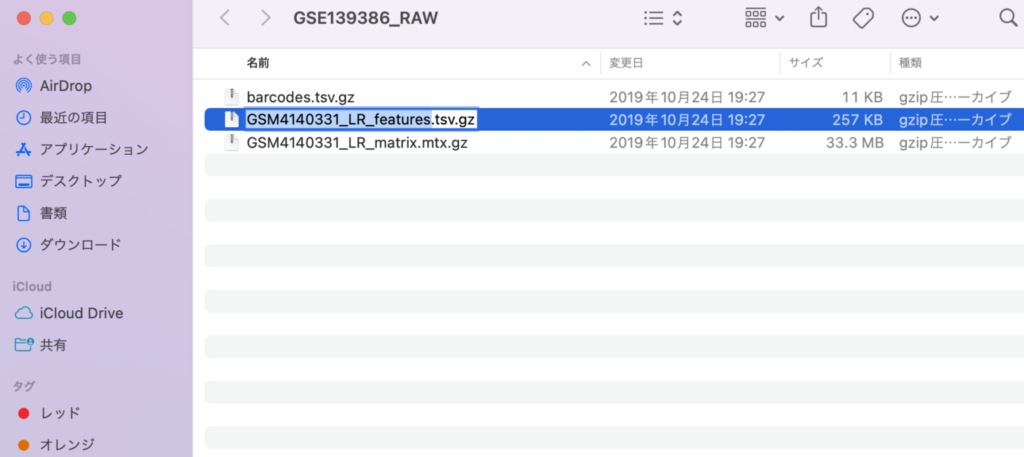
With this, the preparation is complete. The next step is to read the files using Seurat.
When using Seurat, RStudio will be utilized, so if you are unfamiliar with how to use RStudio, please refer to the following article for preparation:
【scRNA-seq】 How to Start scRNA-seq Analysis Using Seurat (Part 1)【Seurat】
In RStudio, navigate to the bottom right panel to proceed to the desktop,
- Check the downloaded file
- Click on More
- Choose Set As Working Directory
It’s important to perform this operation correctly, as failing to do so can prevent subsequent code from executing properly.
The necessary code is as follows, starting with loading the library:
install.packages("dplyr")
install.packages("Seurat")
install.packages("patchwork")
library(dplyr)
library(Seurat)
library(patchwork)Use the following code to read the sample and display a violin plot for quality check purposes.
# read the sample
pbmc_v2.data <- Read10X(data.dir = "./GSE139386_RAW/")
# display violinplot
pbmc_v2 <- CreateSeuratObject(counts = pbmc_v2.data, project = "pbmc_v23k", min.cells = 3, min.features = 200)
pbmc_v2[["percent.mt"]] <- PercentageFeatureSet(pbmc_v2, pattern = "^MT-")
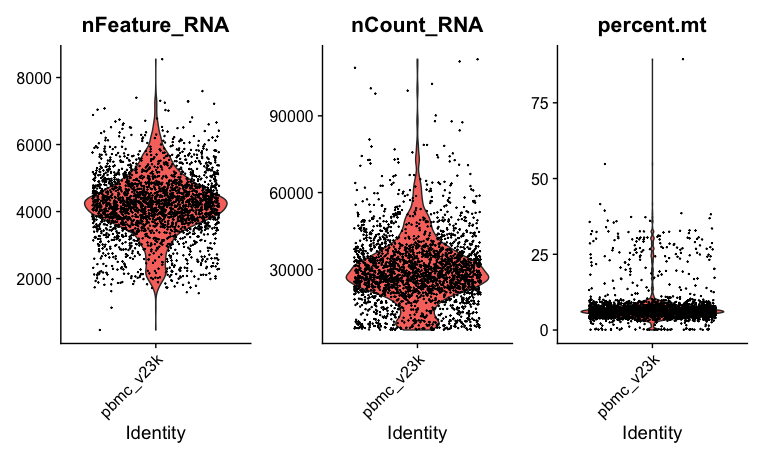
VlnPlot(pbmc_v2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)f you can display a violin plot like the one below, you have succeeded!
The remainder of the analysis is covered in the hands-on articles about Seurat. Please refer to these for guidance on various scRNA-seq visualizations and analyses:
【scRNA-seq】 How to Start scRNA-seq Analysis Using Seurat (Part 1)【Seurat】
【scRNA-seq】 How to Start scRNA-seq Analysis Using Seurat (Part 2)【Seurat】
In Conclusion
How was it? Hopefully, you now have a clear idea of how to find single-cell RNA-seq data related to your research from public databases. Feel encouraged to find data that interests you and give reanalysis a try!

Beginner-Friendly Technical Book on RNA-seq Analysis Using Public Data Available for Sale
¥2,500 → ¥1,500 40%OFF!!
An easy-to-understand guide with abundant screen captures!
Start Dry Research from Your Home PC