
Molecular docking is a computer simulation method that predicts interactions between molecules such as drugs and biomolecules. This article explains the fundamental principles, methods, and specific application examples of molecular docking. It focuses on the LZerD pairwise docking method.
Learning about this topic provides benefits such as effective drug screening and design for drug development, as well as insights into protein interactions and drug optimization. Integrating knowledge from computational and life sciences enables a deeper understanding and broader applications.
Give it a try!
macOS Ventura(13.2.1)
To proceed with this article, please download PyMOL.
What is Molecular Docking?
Molecular docking is a computer simulation technique used to predict how molecules such as drugs and biomolecules (e.g., proteins) interact. It calculates how specific proteins and drugs bind, which parts interact, and predicts the effects of these interactions. This is highly valuable for drug development. Various molecular docking methods are available online. This article focuses on LZerD pairwise docking, which deals with protein-protein docking.
Protein Preparation
First, let’s prepare the protein for docking before analyzing each method. For this example, we will use the leptin protein and its receptor with the Protein Data Bank (PDB) number 8DHA.
Leptin is a protein hormone derived from fat tissue, playing a crucial role in regulating weight and energy balance. It’s released from fat cells into the bloodstream and binds to specific receptors in the hypothalamus, suppressing appetite and regulating metabolism.
Leptin is a protein hormone derived from fat tissue, playing a crucial role in regulating weight and energy balance. It’s released from fat cells into the bloodstream and binds to specific receptors in the hypothalamus, suppressing appetite and regulating metabolism.
The appeal of leptin drugs is their potential to suppress appetite and induce weight loss. This could aid weight management and reduce disease risks for obese or metabolic syndrome patients. Additionally, as leptin is involved in neural circuits controlling appetite, there’s potential for developing new appetite-controlling therapies.
Recent success includes lowering blood sugar using leptin without insulin, offering promise for diabetes treatment.
The leptin and its receptor with the PDB number 8DHA represent only the binding region where they are connected, and the entire structure is registered as 8DH8. To reduce computational complexity in this case, we will utilize the leptin and its receptor with the PDB number 8DHA.
In Pymol, choose File → Get PDB…, enter 8DHA, and display the leptin and its receptor.
Next, use Display → Sequence to show the full sequence.
Select the sequence starting from /C/C/1, which corresponds to leptin.
Then, execute the following code to save the selection as leptin.pdb:
save leptin.pdb, seleSave the receptor part displayed as focus_leptinR.pdb:
save focus_leptinR.pdb, seleAvoid selecting 2-acetamido-2-deoxy-beta-D-glucopyranose.
These files should be saved in Macintosh HD/Users/Your Computer Name.
Protein preparation is now complete.
What is LZerD pairwise docking?
LZerD pairwise docking creates 3D structural models of protein complexes by combining two protein structures (receptor and ligand) as input. It samples potential interaction interface regions and angles to generate thousands of docking structures. These models are then clustered to refine them, and the final ranking of models is determined based on the sum of scores from three scoring functions. This predicts the most likely 3D structure of the protein complex.
Visit the LZerD pairwise docking site.
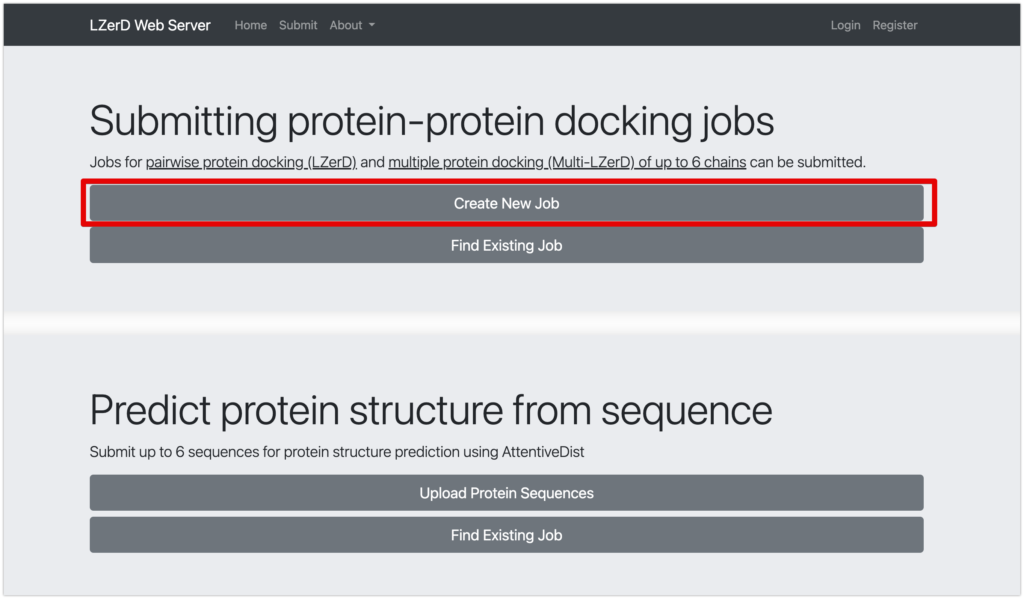
- Select Get Started.
- Under Submitting protein-protein docking jobs, choose Create New Job.*1
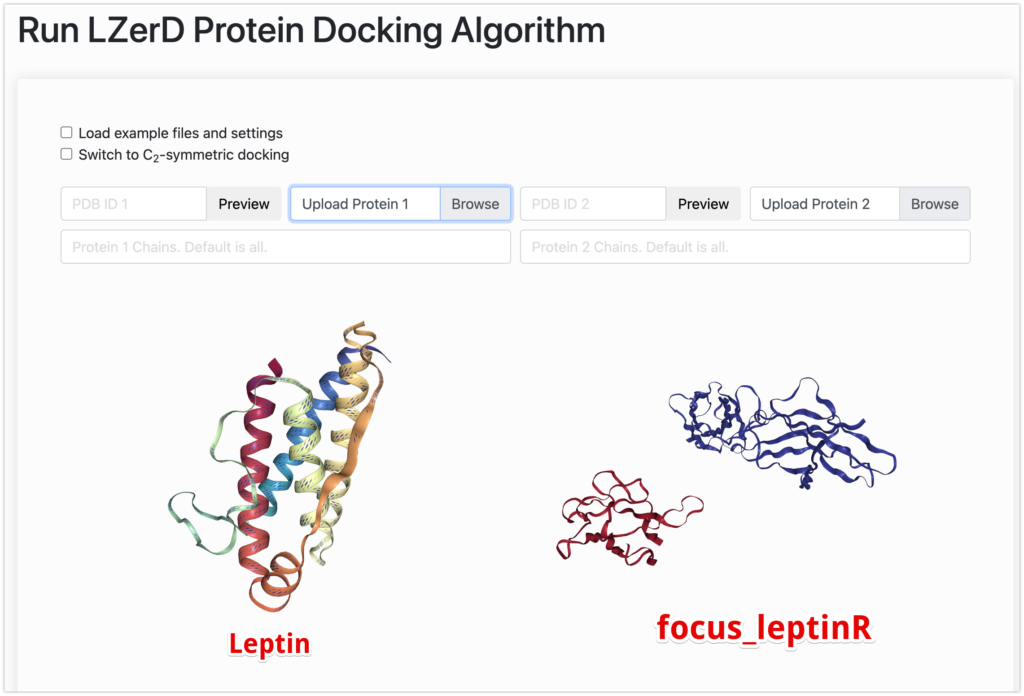
- Upload
leptin.pdbto Upload Protein 1. - Upload
focus_leptinR.pdbto Upload Protein 2.*2 - Provide your email.
- Check Send Email On Job Start.
- Keep other settings as is.
- Submit, and you’ll receive results in your email after a while.
※1
※2
Mechanism of LZerD Pairwise Docking
LZerD (Local 3D Zernike descriptor-based protein Docking) pairwise docking takes two protein structures (receptor and ligand), combines them, and creates a 3D structural model of the protein complex.
This process involves these three main steps:
- LZerD takes the provided structures, samples potential interaction interface regions and angles, and generates thousands of docking structures. Structures with excessive atomic clashes, small interaction regions, or low shape complementarity in the interface area are excluded.
- The generated docking models are clustered using a user-defined clustering cutoff (default is RMSD, 4 Å). This typically reduces the models from thousands to a manageable number.
- The remaining models are ranked using the sum of scores from three scoring functions (DFIRE, GOAP, ITScore). These functions check if atomic interactions in the model resemble distances and angles observed in experimentally determined protein structures.
This way, LZerD pairwise docking efficiently explores and predicts possible 3D structures of protein complexes.
Result
The results are as shown here. It took approximately 1.5 days for the results to be generated.
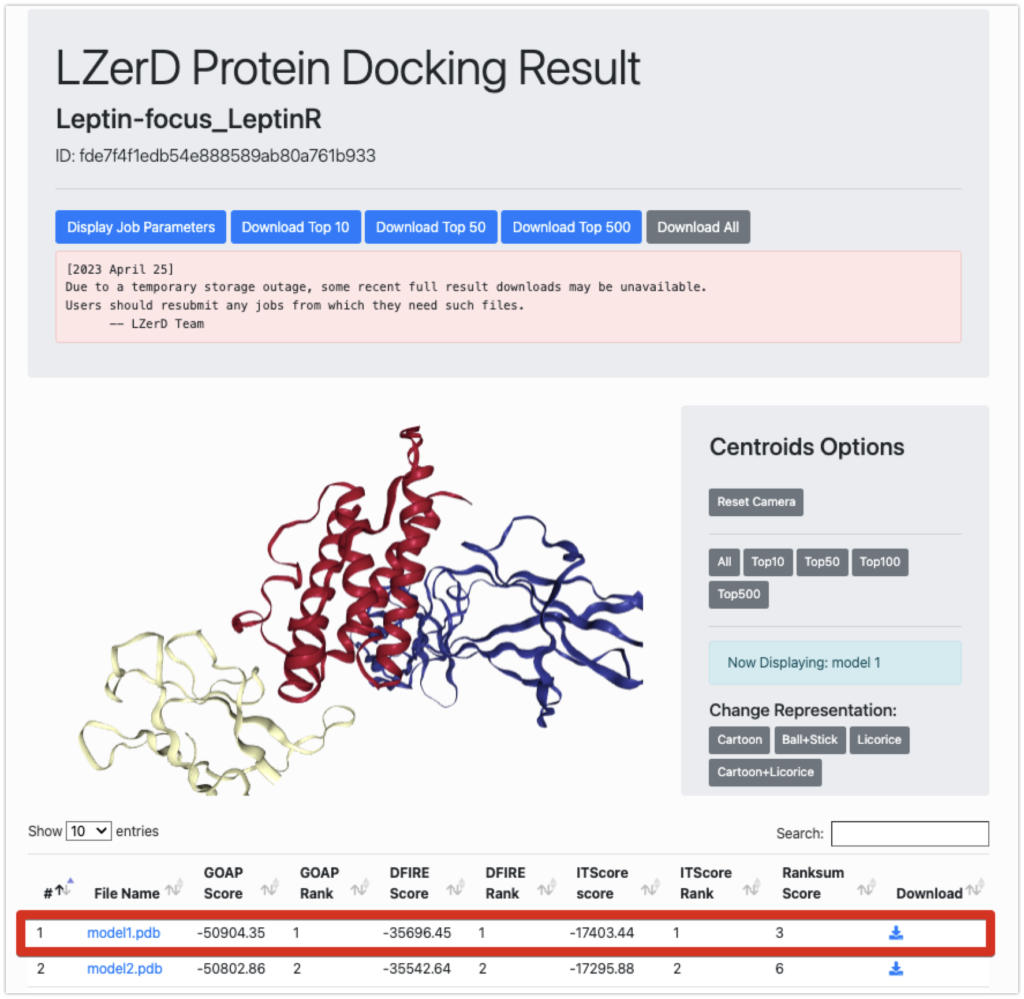
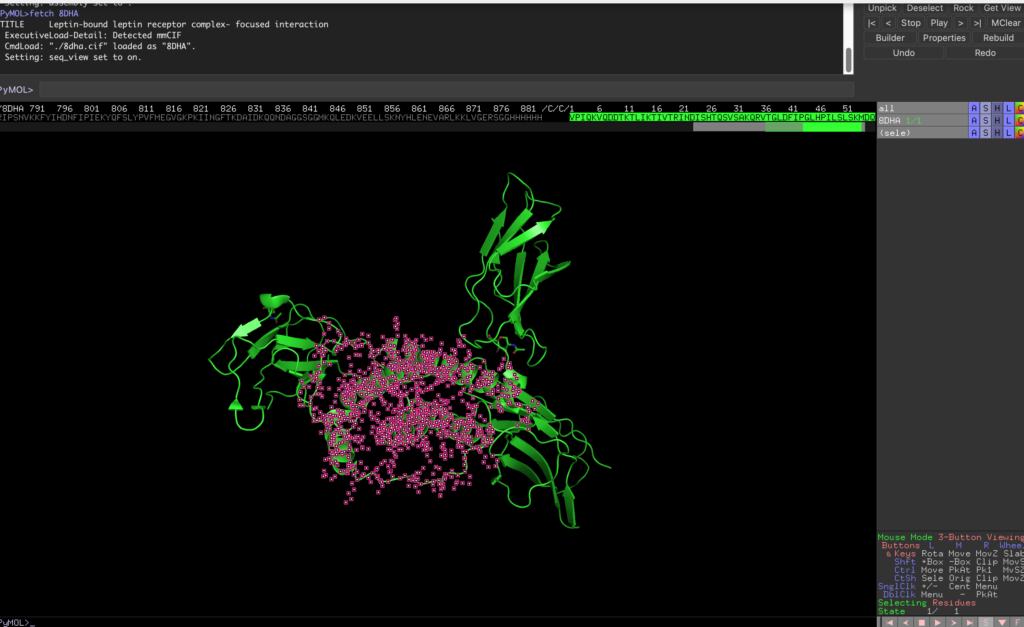
Now, let’s take a look at the lowest-ranking model1.pdb using PyMOL.
Yellow represents Leptin, and green represents focus_leptinR.
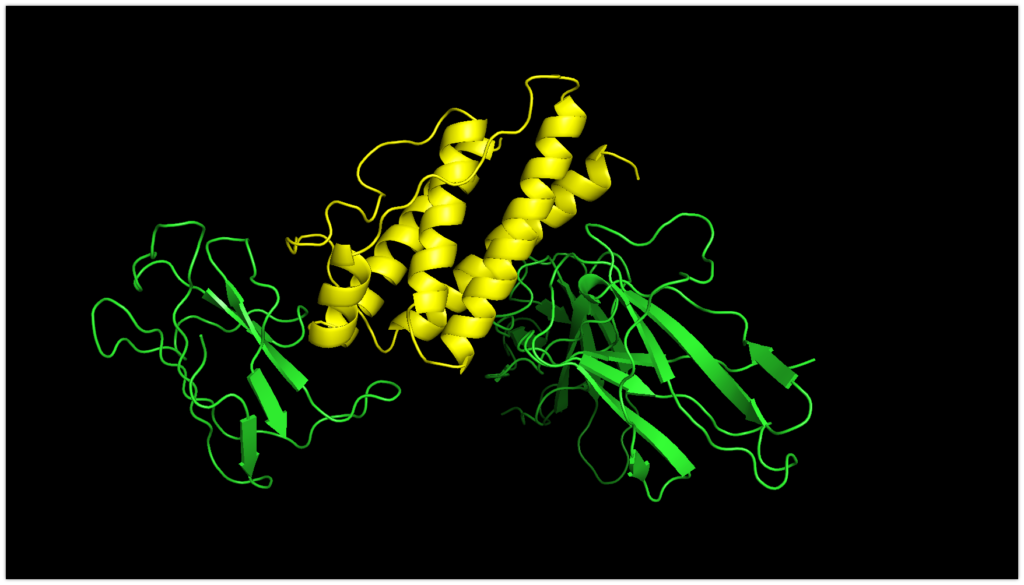
Now, let’s compare it to the original structure.
The magenta color indicates the original structure.
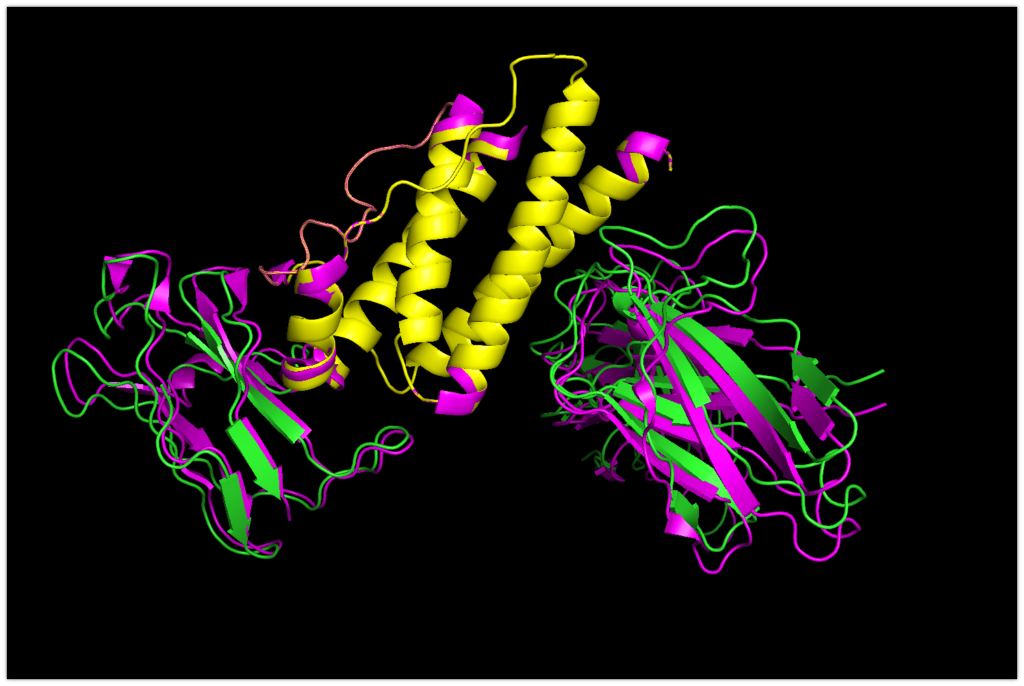
You can see that they are almost bound in a determined position. The accuracy seems to be very promising!
Here, you can notice that focus_LeptinR is slightly misaligned.
Since we used Upload Protein 1 for leptin.pdb and Upload Protein 2 for focus_leptinR.pdb, Upload Protein 1 remained fixed, and Upload Protein 2 was docked with focus_leptinR.pdb.
Therefore, next, let’s try docking by setting Upload Protein 1 to focus_leptinR.pdb and Upload Protein 2 to leptin.pdb.
The result is as shown here.
Yellow-green represents focus_leptinR.pdb used in this docking, and orange represents leptin.pdb that was docked.
The magenta color indicates the original sequence (PDB: 8DHA).

Since focus_leptinR.pdb is not misaligned. It seems that fixing Upload Protein 1 and docking Upload Protein 2 was the correct approach. Nonetheless, it’s surprising to see that leptin.pdb has been docked with such high accuracy.
In Conclusion
How was the information? Today, we discussed LZerD pairwise docking as part of protein-protein docking methods. This method can be easily tried using a server, so I encourage everyone to give it a try. In previous articles, we covered ClusPro and PatchDock, but the principles here are slightly different, and this method demonstrates high accuracy in the case of the presented proteins.
Furthermore, with the LZerD Web Server:
Multi-LZerD Multi-Chain Docking: A method that takes input from three or more protein structures and combines them all to create complex structures.
AttentiveDist Single-Chain Protein Structure Prediction: A method that takes individual protein sequences as input and predicts their structures anew.
These are also possible. If you’re interested, please give them a try!