

本記事は、タンパク質複合体におけるタンパク質間相互作用の結合親和性を予測するPRODIGYについての記事です。
今回は前回の記事で作成した環状ペプチドとタンパク質の複合体を例に、相互作用を予測する過程をご紹介します。
【この記事のまとめ】
タンパク質複合体の立体構造データ(PDB形式)から、結合自由エネルギー()や解離定数()を迅速かつ高精度に予測したい研究者や学生に向けた実践ガイドです。オンラインツール「PRODIGY」を活用し、環境構築不要のWeb版から、大量データ処理に適したコマンドライン(CLI)版までの具体的な操作手順を網羅しています。
- PRODIGYの予測原理: 界面接触残基(ICs)の数と質、および非界面残基(NIS)の性質を指標とする回帰モデルにより、物理化学的計算よりも軽量かつ高速な解析を実現。
- Web版とCLI版の使い分け: ブラウザから数クリックで結果を得る手順と、WSL(Ubuntu)環境でPythonパッケージを用いて一括処理を行う方法の両方を詳しく解説。
- 解析結果の正確な読み方: 出力される$\Delta G$(負に大きいほど強結合)や(モル濃度)、接触残基の種類別カウントなど、各指標の意味と評価のポイントを明示。
この記事を読むことで、複雑なシミュレーション環境を整えることなく、独自のタンパク質・ペプチド複合体モデルの結合強度を客観的な数値で評価できるようになります。
・ブラウザでの利用
Windows 11, Microsoft Edge バージョン 139.0.3405.86 (公式ビルド) (64 ビット)
・コマンドラインでの利用
Windows 11, WSL2(Ubuntu 20.04), Python3.9(Micromamba 管理),bash(Micromamba 初期化済)

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


PRODIGYとは?
PRODIGY(PROtein binDIng enerGY prediction)は、タンパク質間相互作用における結合親和性を予測するためのツールです。
タンパク質–タンパク質複合体の立体構造から、その結合親和性(binding affinity)を高精度に予測し、ギブス自由エネルギーや解離定数 Kdとして親和性を計算するツールで、ブラウザからもコマンドラインからも使用することができます。
こちらのツールは、主にタンパク質–タンパク質間相互作用に対応していますが、拡張版としてPRODIGY-LIG(タンパク質と小分子リガンドの場合)や、PRODIGY-CRYSTAL(生物学的インターフェース vs 結晶インターフェースの分類)も提供されています。ですが、今回はタンパク質–タンパク質間相互作用の予測を用いて、タンパク質-環状ペプチドの結合予測について解説していきます。
PRODIGYのアルゴリズム
PRODIGY のアルゴリズムは、タンパク質複合体の立体構造から結合親和性を予測するために、界面接触というものに着目しています。界面接触とは、(それぞれ異なるタンパク質上にある)分子同士の接触のことを言い、この数が多いほど結合強度が強くなると知られています。PRODIGYではこの界面接触の数と質から結合強度を計算しています。
具体的には、まず、残基間で 5.5Å 以内の原子対を「接触」としてカウントし、それを性質ごとに分類します(荷電・極性・疎水性など)。これにより、複合体界面の相互作用の質と量を数値化できます。さらに、相互作用に関与しない残基表面(NIS: Non-Interacting Surface)の特徴も加えることで、単に接触数だけでは捉えられない安定性の要因を補正します。最終的に、これらの特徴量を説明変数とする回帰モデルにより、自由エネルギー変化(ΔG)や解離定数(Kd)を予測します。このアプローチは物理化学的計算より軽量で、かつ大規模データから得られた統計的関係を利用するため、高速かつ精度の高い予測を実現しています。
PRODIGYの利用法(ブラウザからの利用)
PRODIGY はコマンドラインからも利用が可能ですが、 Web サーバーとしても公開されているため、主要なブラウザ(Google Chrome, Firefox, Safari, Microsoft Edge など)からも利用可能です。
ブラウザからの利用は、多くの立体構造を評価するのには向きませんが、とても簡単にPRODIGYを利用できます。(一方、コマンドラインからの利用は一度に多くの立体構造を評価する際に便利です。コマンドラインからの利用法は後半で紹介します。)
環境構築
ブラウザからの利用は特別な環境は不要で、すぐに使用できます。(筆者は Microsoft Edgeで利用。)
注意点があるとすれば、JavaScript を有効にしておく必要があるようです。もしも無効になっている場合は有効化しておきましょう。
PRODIGYを使った環状ペプチド-タンパク質の結合予測の実行
では、実際にPRODIGYを用いて環状ペプチド-タンパク質結合エネルギーの予測を行っていきます。
今回は、以前の記事でAutodock CrankPepで使った環状ペプチドとタンパク質の立体構造ファイル(example1_omm_rescored_out.pdb)を例に行なっていきます。
- まずはPRODIGYのサイトへ。
- サイト中央の以下の箇所に移動。
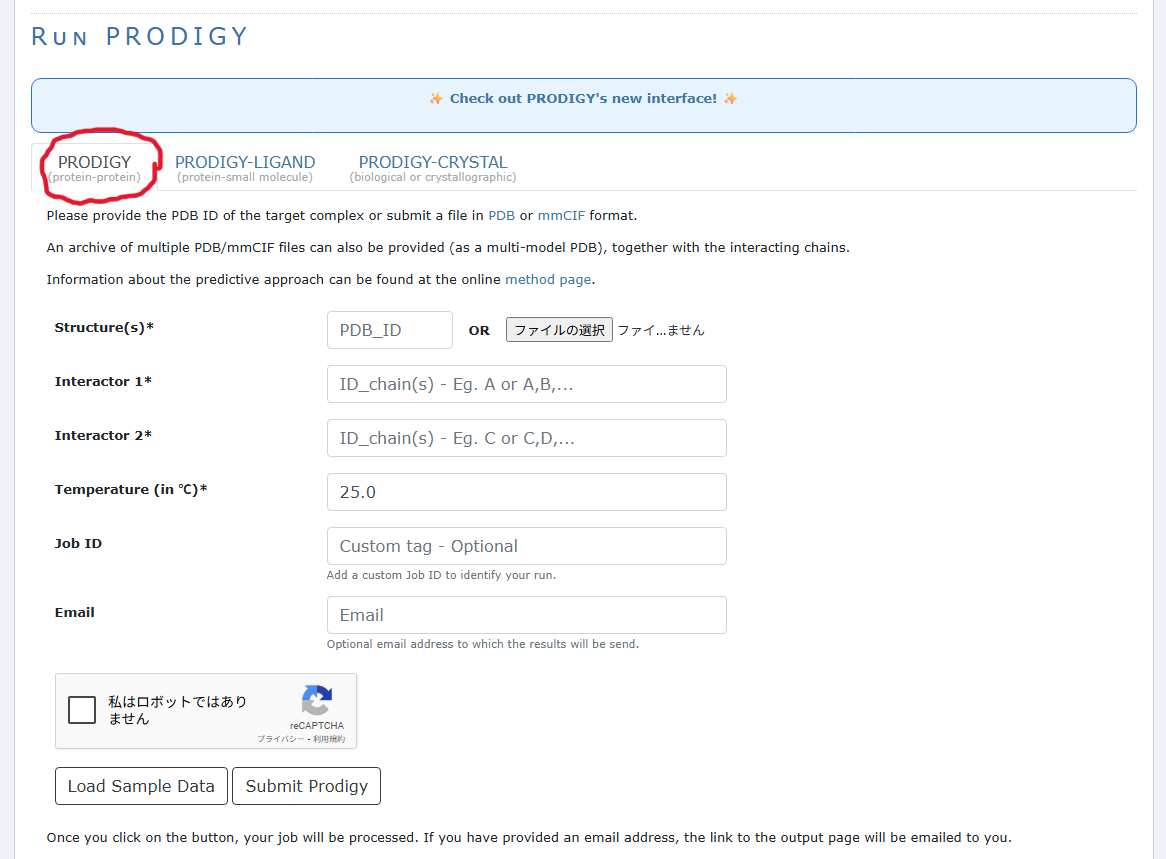
- 上記で赤枠でmarkしたタブでPRODIGY (protein-protein)を選択していることを確認し、Structure(s)から環状ペプチドとタンパク質の立体構造ファイル(example1_omm_rescored_out.pdb)を選択。
- アップロードした PDB/mmCIF ファイルには複数のタンパク質が含まれています。その中から 相互作用を評価したいタンパク質ペアを Interactor 1 と Interactor 2 に指定します。 今回はexample1_omm_rescored_out.pdbの中身に併せて、Interactor 1 にB、 Interactor 2 にZを入力します。 例:もし Chain A と Chain B が結合を予測するなら、Interactor 1 = A、Interactor 2 = B。今回利用したファイルには2つのタンパク質しか含まれませんが、3つ以上のタンパク質が含まれているファイルを選択した場合でも、ここで予測したいペアをしていることが出来ます。
- Temperatureでは結合強度を予測するうえで、どの温度条件での結合強度を予測するかを設定できます。今回は25℃を入力しておきます
- Job IDにはサーバーに投げるjobの名前を入力でき、Emailにメールアドレスを入力しておくと結果がメールにも転送されます。
- 私はロボットではありません。のチェックボックスに入力し、Submit Prodigyをクリック。
実行結果
2~3分ほどで以下のような結果が返ってきます。
今回アップロードしたファイルには受容体タンパク質と環状ペプチドの結合の複合体のモデルが5つ含まれていたので、その5つのモデルそれぞれの結合の強さが計算されました。
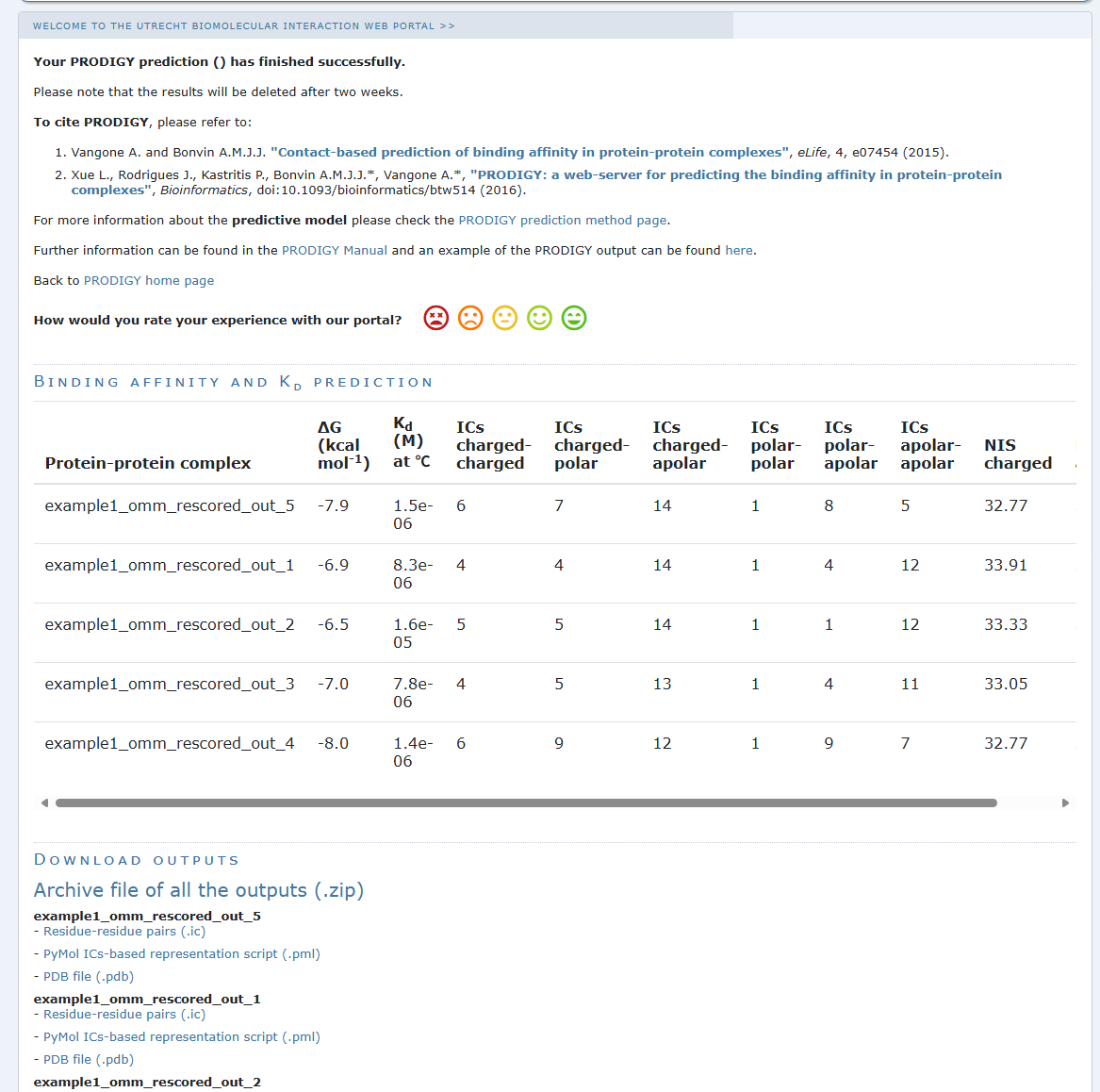
- 表の各列の意味
-Protein-protein complex
解析したファイル名。複数モデル(1〜5)をアップロードしたので、それぞれ行に分かれています。
-ΔG (kcal mol⁻¹)
結合自由エネルギー変化(binding free energy)の予測値。
値が 負(小さいほど強い結合) になるほど、結合親和性が高いことを意味します。
例:-7.9 kcal/mol → 強い相互作用が予測される。
-Kd (M) at °C
平衡解離定数(dissociation constant)の予測値。
単位はモル濃度(M)。
値が 小さいほど強い結合を意味します。
例:1.5e-06 M(マイクロモルオーダー) → 比較的強いタンパク質間結合。
-ICs charged-charged
界面での「荷電残基–荷電残基」の接触数。
-ICs charged-polar
「荷電残基–極性残基」の接触数。
-ICs charged-apolar
「荷電残基–疎水性残基」の接触数。
-ICs polar-polar
「極性残基–極性残基」の接触数。
-ICs polar-apolar
「極性残基–疎水性残基」の接触数。
-ICs apolar-apolar
「疎水性残基–疎水性残基」の接触数。
疎水性相互作用はタンパク質複合体の安定化に重要。
-NIS charged
界面にある荷電残基の割合を示す指標(Normalized Interface Score)。
表のΔGやKdを見てみると、モデル5やモデル4の値が小さく、強い結合であると予測されたようです。逆にモデル1や2の結合はそれよりも弱いと予測されました。
結果の画面の下部から結果をダウンロードすることもできます。
コマンドラインを使った方法
実用的には、一度に大量の立体構造を評価することが多く、その際はコマンドラインを利用してPRODIGYを利用するのが良いでしょう。
先ほどと同様に環状ペプチドとタンパク質の立体構造ファイル(example1_omm_rescored_out.pdb)を例に行なっていきます。
こちらを参考に進めていきます。
環境構築
環境構築をしていきます。今回はWindowsのWSLを利用していきます。WSLの利用方法は前回の記事に記載してあるので、そちらを参考にしてください。
WSLで以下のコードを実行して、環境構築をしていきます。
# micromamba をインストールするディレクトリを作成
mkdir -p ~/micromamba_env
# micromamba をインストール(-b: バッチモード, -p: インストール先指定)
curl -Ls https://micro.mamba.pm/install.sh | bash -s -- -b -p ~/micromamba_env
# bash 設定ファイルを読み込み直す(PATH 設定などを反映)
source ~/.bashrc
# PRODIGY用の環境を作成
micromamba create -n prodigy_env python=3.9 -y
# 作成した環境をアクティベート
micromamba activate prodigy_env
# PRODIGYとその他に必要なパッケージのインストール
micromamba install -n prodigy_env -c conda-forge freesasa freesasa-python -y
pip install --upgrade pip
pip install prodigy-prot biopythonコードの詳細説明
mkdir -p ~/micromamba_env curl -Ls https://micro.mamba.pm/install.sh | bash -s -- -b -p ~/micromamba_env source ~/.bashrc
micromambaを使った環境構築をしていきます。これらはそのmicromambaをインストールするコマンドです。すでにインストールしている場合は、不要です。
micromamba create -n prodigy_env python=3.9 -y
micromambaで「prodigy_env」という名前の仮想環境を作成しています。
micromamba create: 新しい環境を作成するコマンドn prodigy_env: 環境の名前を prodigy_env とするpython=3.9: Python 3.9 をインストール(PRODIGYはPython 3.7以上が必要です。)y: ユーザー確認なしで自動的に進める
👉 これで prodigy_env という独立した環境ができ、Python 3.9 が入ります。
micromamba activate prodigy_env
micromambaで作られた「prodigy_env」という名前の仮想環境(パッケージや設定がまとまった作業用スペース)を「有効化(activate)」します。
これを実行すると、その環境の中に入って、その環境にインストールされたツールやライブラリが使える状態になります。
micromamba install -n prodigy_env -c conda-forge freesasa freesasa-python -y
micromambaでBSA計算に必要なパッケージ(freesasa)をインストールします。
micromamba install: 環境にパッケージを追加、micromambaでインストールすることで他の環境を汚さずに、指定した環境のみにインストールすることができます。-n prodigy_env: 追加先を prodigy_env に指定-c conda-forge: conda-forge チャンネル(オープンソースのパッケージリポジトリ)から取得
pip install --upgrade pip
インストールに使用する関数pipをアップグレードしておきます。pip install prodigy-prot biopython
pipでPRODIGY本体(prodigy-prot)とそれを動かすのに必要なパッケージ(biopython)をインストール
PRODIGYの実行
前回作成した立体構造ファイル(example1_omm_rescored_out.pdb)を作業したいフォルダにコピーします。(私はdocuments下に作ったPRODIGYというフォルダに入れました)
コピーした後は、以下のコマンドで作業したいフォルダに移動します。/documents/PRODIGYは、ご自身が作業したいフォルダの名前に合わせて変更して下さい。
cd /mnt/c/users/(あなたのWondowsのユーザー名)/documents/PRODIGYそして以下を実行します。
# ファイルを立体構造モデル1つ1つにバラバラにする
awk ' /^MODEL/ { if (out) close(out); count++; out = sprintf("model_%03d.pdb", count) } { print > out } /^ENDMDL/ { close(out) }
' example1_omm_rescored_out.pdb
# PRODIGY を実行
prodigy model_001.pdb
# いくつかのオプションをつけて実行
prodigy model_001.pdb --distance-cutoff 5 --acc-threshold 20 --temperature 25 --contact_list --pymol_selection --selection B Zコードの詳細説明
awk ' /^MODEL/ { if (out) close(out); count++; out = sprintf("model_%03d.pdb", count) } { print > out } /^ENDMDL/ { close(out) } ' example1_omm_rescored_out.pdb
example1_omm_rescored_out.pdbのファイルには複数の立体構造モデルが含まれていますが、コマンドからPRODIGYを利用する場合、モデルを1つずつ与える必要があります。このコマンドでファイルをモデルごとに分割しておきます。
prodigy model_001.pdb
こちらのコマンドで指定のファイル(model_001.pdb )の結合親和性を予測できます。PDB形式以外にもmmFIC形式でも実行できます。また、
prodigy <directory_with_molecules>
この様にファイル名だけでなく、ディレクトリ名を指定することでディレクトリ内の全てのファイルに対して解析をすることができます。
prodigy model_001.pdb --distance-cutoff 5 --acc-threshold 20 --temperature 25 --contact_list --pymol_selection --selection B Z
また、コマンドラインから使用すると、この様にオプションを利用してより詳細な解析ができます。
利用可能なオプションの説明は以下の通りです:
-distance-cutoff DISTANCE_CUTOFF相互作用をカウントする際の距離カットオフを指定。-acc-threshold ACC_THRESHOLDBSA(Buried Surface Area)解析の際のアクセスしやすさの閾値設定。-temperature TEMPERATURE解離定数(Kd)予測時に使用する温度(℃)を指定。-contact_listインターモレキュラー接触のリストを出力。-pymol_selectionPyMOL 用の選択・ハイライトスクリプトを出力。-q, --quiet出力を結合親和性値のみ等の簡素な形に抑える。prodigy -q input_dir/こちらのコマンドのように複数のファイルを一括で処理する際に利用すると便利です。-selection A B [A,B C ...]特定のチェーン間でのみ接触計算したい場合に使用。 コロンで区切られた複数チェーンのグループとして扱えます。-selection A B→ チェーン A と B の間のみ-selection A,B C→ A と C、B と C の接触計算-selection A B C→ A–B、B–C、A–C すべて計算
実行結果
prodigy model_001.pdb
こちらを実行させたところ、以下のような結果になりました。
(prodigy_env) ~~~~ $ prodigy model_001.pdb
[+] Parsed structure file model_001 (2 chains, 143 residues)
[+] No. of intermolecular contacts: 39
[+] No. of charged-charged contacts: 4.0
[+] No. of charged-polar contacts: 4.0
[+] No. of charged-apolar contacts: 14.0
[+] No. of polar-polar contacts: 1.0
[+] No. of apolar-polar contacts: 4.0
[+] No. of apolar-apolar contacts: 12.0
[+] Percentage of apolar NIS residues: 36.52
[+] Percentage of charged NIS residues: 33.91
[++] Predicted binding affinity (kcal.mol-1): -6.9
[++] Predicted dissociation constant (M) at 25.0˚C: 8.3e-06ブラウザ版のmodel1と同じ結果が得られました。
最後に
いかかでしたでしょうか。
PRODIGYはwebから使えることもあり、簡単に使用することが出来ました!
前回の記事では結合力とドッキングポーズを評価しましたが、今回はそれをもとにもうちょっときちんとした評価指標ΔGとKdで立体構造を評価していきました。
実際には、こちらの論文https://chemrxiv.org/engage/chemrxiv/article-details/63c7a4da23c13b2e4813aa5bのように両方を相互的にみて最終的な判断をすることが多いようです。
参考文献
Apache License v2.0
PRODIGY: a web server for predicting the binding affinity of protein–protein complexes
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

