

この記事では、GROMACSを用いて、外部から特定の力を加えて、分子の動きを意図的に誘導するSteered Molecular Dynamics(SMD)を行い、それぞれのポイント初期構造として、バイアスをかけてシミュレーションをし、エネルギー変化を見るUmbrella samplingというものを紹介します。in silico創薬ではこれを利用し、結合エネルギーや膜透過性の評価などが行われています。ここでは一連の動作を通して、アミロイドベータ(Aβ)ペプチドがその集合体の線維から離れる時の自由エネルギーを計算しています。
【この記事のまとめ】
GROMACSを用いたMDシミュレーションにおいて、タンパク質とリガンドなどの分子間相互作用における自由エネルギー変化(PMF:平均力ポテンシャル)を定量化する「アンブレラサンプリング(Umbrella Sampling)」の実装手順を解説します。
- プル・シミュレーションによる構造生成: 反応座標(分子間距離)に沿ってリガンドを強制的に引き離す「Steered MD」を行い、アンブレラサンプリングの初期構造となる一連の「ウィンドウ(スナップショット)」を作成する手法を提示。
- 調和ポテンシャルによるサンプリング: 各ウィンドウに対して調和ポテンシャル(バイアス)を適用し、通常ではサンプリングが困難な高エネルギー状態(結合・解離の遷移過程)を重点的にシミュレーションする設定方法を詳説。
- WHAM法を用いた自由エネルギーの再構築:
gmx whamコマンドを使用し、各ウィンドウから得られた偏りのあるサンプリング結果を統合・解析して、反応座標に沿った自由エネルギープロファイルを算出・可視化するプロセスを紹介。
この記事を読むことで、単なる結合ポーズの観察に留まらず、結合親和性の強さや解離に伴うエネルギー障壁を数値として正確に把握し、創薬研究における分子設計の根拠を強化できるようになります。
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


Umbrella Samplingの概要と目的
Umbrella Samplingは、分子がめったに起こさないような状態変化(たとえば、タンパク質同士が離れるなど)を詳しく調べるための計算手法です。通常の分子動力学(MD)では、エネルギー的に安定な状態にとどまりやすく、重要な変化が起こる確率が低いため、十分な情報が得られません。
たとえば、タンパク質AとBがくっついている状態から、少しずつ離れていく様子を観察したいとき、まずAとBを少しずつ引き離すSteered Molecular Dynamics(SMD)などのシミュレーションで、さまざまな距離の構造を取得します。その中から、0.5nm、0.7nm、0.9nmなど、特定の距離ごとに「窓(window)」を設定し、それぞれの距離でバネのような力をかけてMDシミュレーションを行います。これにより、その距離での分子の振る舞いをしっかり観察できます。
最後に、WHAMという手法を用いて全ての窓の情報を統合し、自由エネルギー変化(Potential of Mean Force: PMF)を計算します。
ここではGROMACSのumbrella Samplingのチュートリアルを例に解説していきます。
一連の動作を通して、アミロイドベータ(Aβ)ペプチドがその集合体から離れる時の自由エネルギーを計算してみましょう
若干古いチュートリアルなので、所々warningが出ます。本来なら修正をすべきですが、とりあえずはwarningは無視して行います。
Umbrella samplingについて、よく理解できなかった方は以下の動画をみるとよさそうです。
アンブレラサンプリング法を用いた自由エネルギーの算出【計算化学、理論化学】
MD 初心者の方は以下の記事を参考にMDの基礎を行うとよいです。
環境構築
GROMACSによるMD simulation(分子動力学シミュレーション)のためのWindows環境の構築をやってみた
タンパク質のみのMD
【GROMACS】MD simulationを用いたタンパク質の安定化【タンパク質デザイン】【In silico創薬】 – LabCode
低分子ーリガンドのMD
GROMACSでリガンド-タンパク質 MDsimulation(分子動力学シミュレーション)を論文に沿ってやってみた
Umbrella Samplingの流れ
- Generate Topology (トポロジー生成):シミュレーションを行う分子(今回の場合はタンパク質)の原子の種類、結合、電荷などの情報を含むトポロジーファイルを生成します。
- Define Unit Cell (単位格子の定義)、Solvate and Add Ions (溶媒追加とイオン付加):シミュレーション空間(ボックス)のサイズを定義し、タンパク質を水分子で囲み、生体内の環境に近い状態にするためにイオンを追加します。
- Minimization and Equilibration (エネルギー最小化と平衡化):シミュレーションを開始する前に、系のエネルギーを最小化し、安定な状態に平衡化します。これにより、初期構造に起因する不自然な挙動を防ぎます。
- Generate Configurations (コンフィギュレーション生成):アンブレラサンプリングの各「窓(window)」でシミュレーションを行うための初期構造を生成します。これは通常、タンパク質を引き離す「引き抜き(pulling)」シミュレーションによって行われます。
- Umbrella Sampling (アンブレラサンプリング):各窓でシミュレーションを実行し、それぞれの窓で異なる拘束ポテンシャル(アンブレラポテンシャル)を適用して、特定の反応座標に沿ったサンプリングを強化します。
- Data Analysis (データ解析):アンブレラサンプリングで得られたデータを用いて、反応座標に沿った自由エネルギープロファイル(Potential of Mean Force, PMF)を計算します。
最終的に以下のようなグラフができます。

このグラフは、**Aβ42プロトフィブリルからペプチドが離れていく際の、必要な「エネルギーの坂道」**を示しています。
- X軸 (距離):ペプチドがプロトフィブリルからどれだけ離れたか。
- Y軸 (エネルギー):その距離まで離れるのに必要なエネルギー。
グラフが示すこと:
- ペプチドが約0.5nmから3.7nm離れるまでは、**エネルギーが急激に上昇(約48 kcal/mol必要)**します。これは、この分離には大きな抵抗(高い坂)があることを意味します。
- 約3.7nmを超えると、エネルギーはそれ以上上がらず横ばいになります。これは、完全に分離し、それ以上離れてもエネルギーの変化がほとんどない状態(坂を登りきった状態)になったことを示します。
つまり、このグラフはAβ42プロトフィブリルからペプチドを分離させるには、約48 kcal/molのエネルギーが必要であることを示しています。
では次からこれをやっていきましょう。
Umbrella Samplingの実行
Generate Topology (トポロジー生成)
まず、GROMACSでシミュレーションを行うために必要な分子の情報を定義する トポロジーファイル を生成します。これは、各原子の種類、それらの間の結合(結合長、結合角、二面角)、そして原子が持つ電荷など、分子の構造と相互作用に関するすべての情報を含みます。
サンプルPDBファイルの準備
まずお好きな階層に作業ディレクトリを作りましょう。以下はターミナルで行ってください。
mkdir umbrella_sampling
cd umbrella_sampling
# ここに以下で得られる2BEG_model1_capped.pdbファイルをダウンロードして配置してください次に、シミュレーション対象となるタンパク質のPDB (Protein Data Bank) ファイルを用意します。今回は、まずチュートリアルで指定されたPDBファイルをダウンロードします。
このサイトの以下の部分をクリックして、ダウンロードしてください。

PDB ID: 2BEGのタンパク質は、アルツハイマー病の原因とされるアミロイドベータ(Aβ)ペプチド42残基(Aβ42)が集合して形成された、線維(フィブリル)の前段階であるプロトフィブリルの3次元構造 です。N末端がアセチル化されており、これはシミュレーションで安定性を高めるためによく行われる修飾です。
ダウンロードしたPDBを、作業ディレクトリ umbrella_sampling に配置します。
次に以下を実行してください。
gmx pdb2gmx コマンドは、PDBファイルからGROMACS形式の構造ファイル(.gro)とトポロジーファイル(.top、.itp)を生成します。
gmx pdb2gmx -f 2BEG_model1_capped.pdb -ignh -ter -o complex.grogmx pdb2gmx コマンドは、PDBファイルからGROMACS形式の構造ファイル(.gro)とトポロジーファイル(.top、.itp)を生成します。
f 2BEG_model1_capped.pdb: 入力PDBファイルを指定します。ignh: PDBファイルに記載されている水素原子を無視します。GROMACSのフォースフィールドは、通常、水素原子を自動的に追加するため、このオプションを使用します。ter: N末端およびC末端の処理について対話形式で尋ねるように指定します。o complex.gro: 出力するGROMACS形式の構造ファイル名を指定します。
コマンドを実行すると、いくつか質問が表示されます。
shizuku@DESKTOP-5I5GHRA:~/umbrella_sampling$ gmx pdb2gmx -f 2BEG_model1_capped.pdb -ignh -ter -o complex.gro :-) GROMACS - gmx pdb2gmx, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/umbrella_sampling
Command line: gmx pdb2gmx -f 2BEG_model1_capped.pdb -ignh -ter -o complex.gro
Select the Force Field:
From '/usr/local/gromacs/share/gromacs/top': 1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003) 2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995) 3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996) 4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000) 5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006) 6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010) 7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002) 8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins) 9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)多くの選択肢が表示されますが、今回は 「13: GROMOS96 53a6 force field」 を選択します。フォースフィールドは、原子間の相互作用を記述する数学的なモデルであり、シミュレーションの精度に大きく影響します。GROMOS96 53a6はタンパク質シミュレーションによく用いられるフォースフィールドの一つです。
次に以下が出てきます。
Using the Gromos53a6 force field in directory gromos53a6.ff
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/watermodels.dat
Select the Water Model: 1: SPC simple point charge, recommended 2: SPC/E extended simple point charge 3: None次に、水分子のモデルを選択します。今回は 「1: SPC simple point charge, recommended」 を選択します。SPCは広く使用されている水モデルの一つです。
最後にPDBファイルからタンパク質を読み込む際に、N末端とC末端の処理について尋ねられます。指示に従い、N末端には 「None (3)」、C末端には 「COO- (0)」 を選択します。
going to rename gromos53a6.ff/aminoacids.r2b
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/aminoacids.r2b
Reading 2BEG_model1_capped.pdb...
Read 'Gallium Rubidium Oxygen Manganese Argon Carbon Silicon t= 0.00000', 920 atoms
Analyzing pdb file
Splitting chemical chains based on TER records or chain id changing.
There are 5 chains and 0 blocks of water and 135 residues with 920 atoms chain #res #atoms 1 'A' 27 184 2 'B' 27 184 3 'C' 27 184 4 'D' 27 184 5 'E' 27 184
All occupancies are one
All occupancies are one
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/atomtypes.atp
Reading residue database... (Gromos53a6)
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/aminoacids.rtp
Using default: not generating all possible dihedrals
Using default: excluding 3 bonded neighbors
Using default: generating 1,4 H--H interactions
Using default: removing proper dihedrals found on the same bond as a proper dihedral
Using default: removing proper dihedrals found on the same bond as a proper dihedral
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/aminoacids.hdb
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/aminoacids.n.tdb
Opening force field file /usr/local/gromacs/share/gromacs/top/gromos53a6.ff/aminoacids.c.tdb
Processing chain 1 'A' (184 atoms, 27 residues)
Identified residue ACE1 as a starting terminus.
Identified residue ALA27 as a ending terminus.
9 out of 9 lines of specbond.dat converted successfully
Select start terminus type for ACE-1 0: NH3+ 1: NH2 2: None2; “None” を選択します。
Start terminus ACE-1: None
Select end terminus type for ALA-27 0: COO- 1: COOH 2: None0: COO- を選択します。
何回か出てくるので、N-terminiには “None” , C-terminiには”COO-” を選択してください。
以上のコマンドの実行により、以下のファイルが生成されます。
complex.gro: GROMACS形式の構造ファイルtopol.top: メインのトポロジーファイル。シミュレーションで必要なすべての.itpファイルをインクルードします。topol_Protein_chain_A.itp,topol_Protein_chain_B.itpなど: 各タンパク質鎖のトポロジー情報が含まれるファイル。
次にアンブレラサンプリングの引き抜きシミュレーションで、チェーンBを不動の参照点として使用する ために、topol_Protein_chain_B.itp ファイルを修正します。この修正により、チェーンBに位置拘束(position restraint)をかけることができるようになります。
テキストエディタで topol_Protein_chain_B.itp を開き、ファイルの最後に以下の行を追加します。
;
; File 'topol_Protein_chain_B.itp' was generated
; By user: unknown (1000)
; On host: DESKTOP-5I5GHRA
; At date: Thu Jun 12 19:44:37 2025
;
; This is a include topology file
;
; Created by:
; :-) GROMACS - gmx pdb2gmx, 2024 (-:
;
; Executable: /usr/local/gromacs/bin/gmx
; Data prefix: /usr/local/gromacs
; Working dir: /home/shizuku/umbrella_sampling
; Command line:
; gmx pdb2gmx -f 2BEG_model1_capped.pdb -ignh -ter -o complex.gro
; Force field was read from the standard GROMACS share directory.
;
[ moleculetype ]
; Name nrexcl
Protein_chain_B 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 28 ACE rtp ACE q 0.0 1 CH3 28 ACE CA 1 0 15.035 2 C 28 ACE C 2 0.45 12.011
~(中略)~ 213 211 218 214 2 gi_2 213 216 215 214 2 gi_2 218 213 220 219 2 gi_1 220 218 222 221 2 gi_1 222 220 224 223 2 gi_2 224 222 226 225 2 gi_1
; Include Position restraint file
#ifdef POSRES_B (ここを修正)
#include "posre_Protein_chain_B.itp"
#endifこの #ifdef POSRES_B のブロックを追加することで、後に .mdp ファイルで define = -DPOSRES_B を設定した場合にのみ、posre_Protein_chain_B.itp ファイルが読み込まれ、チェーンBに位置拘束が適用されるようになります。これにより、他のチェーンには拘束がかからない状態で、チェーンAを引き抜くことが可能になります。
Define Unit Cell, Solvate and Add Ions
タンパク質分子は通常、真空中ではなく、水溶液中で機能します。そのため、シミュレーションを行う際には、タンパク質を適切なサイズの水ボックスに入れ、生理的なイオン濃度を再現する必要があります。
まず以下を実行し、gmx editconf コマンドを使って、シミュレーションの対象となる空間(単位セル、または ボックス と呼ばれます)のサイズと形状を定義し、その中心にタンパク質を配置します。
gmx editconf -f complex.gro -o newbox.gro -center 3.280 2.181 2.4775 -box 6.560 4.362 12f complex.gro: 入力となる構造ファイル。o newbox.gro: 出力する新しいボックスに配置された構造ファイル。center 3.280 2.181 2.4775: タンパク質の重心をボックスの中心に配置します。指定された座標は、ボックスの半分のサイズです。box 6.560 4.362 12: ボックスのX, Y, Z方向のサイズをナノメートル(nm)単位で指定します。
次に
gmx solvate コマンドを実行し、定義されたボックス内に水分子を充填します。
gmx solvate -cp newbox.gro -cs spc216.gro -o solv.gro -p topol.topcp newbox.gro: 水を充填する対象のタンパク質構造ファイル。cs spc216.gro: 使用する水モデルの構造ファイル(GROMACSに標準で含まれています)。o solv.gro: 水が充填された新しい構造ファイル。p topol.top: 水分子のトポロジー情報を追加するために、メインのトポロジーファイルを更新します。
水分子を充填した後、系の電荷を中和し、生理的なイオン濃度を再現するためにイオンを追加します。
まず、イオン化のための設定ファイル ions.mdp を作成します。
ions.mdp として、以下のファイルを同ディレクトリに作成してください。
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.4 ; Cut-off for making neighbor list (short range forces)
coulombtype = cutoff ; Treatment of long range electrostatic interactions
rcoulomb = 1.4 ; Short-range electrostatic cut-off
rvdw = 1.4 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditionsintegrator = steep: 最急降下法によるエネルギー最小化を行います。これはイオンを追加する前に、追加されたイオンと既存の分子との間で過度な反発がないようにするためです。emtol = 1000.0: エネルギー最小化の収束基準。nsteps = 50000: 最大ステップ数。- その他の設定は、近傍探索や相互作用計算に関するものです。
次に、この .mdp ファイルを使って、イオン化のための実行ファイル(.tpr)を生成します。
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr -maxwarn 1f ions.mdp: 入力とするMDPファイル。c solv.gro: 水が充填された構造ファイル。p topol.top: 更新されたトポロジーファイル。o ions.tpr: 出力する実行ファイル。maxwarn 1: 警告メッセージが1つまでなら処理を続行します。
最後に、gmx genion コマンドで実際にイオンを追加します。
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral -conc 0.1s ions.tpr: イオン化のための実行ファイル。o solv_ions.gro: イオンが追加された新しい構造ファイル。p topol.top: トポロジーファイルを更新し、追加されたイオンの情報を書き込みます。pname NA,nname CL: 正電荷イオン(カチオン)と負電荷イオン(アニオン)として、それぞれナトリウムイオン(NA)と塩化物イオン(CL)を使用することを指定します。neutral: 系の総電荷がゼロになるようにイオンを追加します。conc 0.1: 最終的なイオン濃度を0.1 M(モル)に設定します。
このコマンドの実行中、どのグループにイオンを追加するか尋ねられます。「13: SOL」 (溶媒の水分子のグループ)を選択します。
shizuku@DESKTOP-5I5GHRA:~/umbrella_sampling$ gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral -conc 0.1 :-) GROMACS - gmx genion, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/umbrella_sampling
Command line: gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral -conc 0.1
Reading file ions.tpr, VERSION 2024 (single precision)
Reading file ions.tpr, VERSION 2024 (single precision)
Will try to add 31 NA ions and 21 CL ions.
Select a continuous group of solvent molecules
Group 0 ( System) has 33311 elements
Group 1 ( Protein) has 1130 elements
Group 2 ( Protein-H) has 920 elements
Group 3 ( C-alpha) has 135 elements
Group 4 ( Backbone) has 400 elements
Group 5 ( MainChain) has 540 elements
Group 6 ( MainChain+Cb) has 645 elements
Group 7 ( MainChain+H) has 670 elements
Group 8 ( SideChain) has 460 elements
Group 9 ( SideChain-H) has 380 elements
Group 10 ( Prot-Masses) has 1130 elements
Group 11 ( non-Protein) has 32181 elements
Group 12 ( Water) has 32181 elements
Group 13 ( SOL) has 32181 elements
Group 14 ( non-Water) has 1130 elements
Select a group: Minimization and Equilibration
イオンが追加された後、シミュレーションを開始する前に、系のエネルギーを最小化し、温度と圧力を目標値に平衡化する必要があります。
まず、系の構造的な問題を解消し、原子間の過度な反発などを取り除くために、エネルギー最小化 を行います。これにより、初期構造に起因する不安定な挙動を防ぎます。
以下のminim.mdp ファイルを作成します。
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.4 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.4 ; Short-range electrostatic cut-off
rvdw = 1.4 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditionsintegrator = steep: 最急降下法によるエネルギー最小化を行います。emtol,emstep,nsteps: 最小化の収束基準とステップ数。coulombtype = PME: 長距離静電相互作用の計算にPME (Particle Mesh Ewald) 法を使用します。PMEは周期境界条件を持つ系で正確な計算を行うための標準的な手法です。
このMDPファイルを使って、最小化のための実行ファイル(.tpr)を生成し、シミュレーションを実行します。以下を実行してください。
gmx grompp -f minim.mdp -c solv_ions.gro -p topol.top -o em.tpr -maxwarn -1
gmx mdrun -v -deffnm emgmx grompp:minim.mdp,solv_ions.gro,topol.topを基にem.tprを生成します。maxwarn -1は、すべての警告を無視して処理を続行することを意味します(推奨されませんが、このチュートリアルでは簡略化のために使用)。gmx mdrun:em.tprを使ってエネルギー最小化を実行します。v: 詳細な出力を表示します。deffnm em: 出力ファイルにemというプレフィックスを付けます(例:em.log,em.gro,em.edrなど)。
エネルギー最小化の後、系を目標の温度と圧力に 平衡化 します。これにより、シミュレーションが物理的に意味のある条件で実行されるようになります。平衡化は通常、NPTアンサンブル(定温定圧アンサンブル)で行われます。
npt.mdp ファイルを作成してください。
title = NPT Equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 1000 ; save coordinates every 2 ps
nstvout = 1000 ; save velocities every 2 ps
nstenergy = 1000 ; save energies every 2 ps
nstlog = 1000 ; update log file every 2 ps
; Bond parameters
continuation = no ; Initial simulation
constraint_algorithm = lincs ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
ns_type = grid ; search neighboring grid cels
nstlist = 5 ; 10 fs
rlist = 1.4 ; short-range neighborlist cutoff (in nm)
rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm)
rvdw = 1.4 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = Berendsen ; Weak coupling for initial equilibration
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 310 310 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Berendsen ; Pressure coupling on in NPT, also weak coupling
pcoupltype = isotropic ; uniform scaling of x-y-z box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure (in bar)
compressibility = 4.5e-5 ; isothermal compressibility, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = yes ; Velocity generation is on
gen_temp = 310 ; temperature for velocity generation
gen_seed = -1 ; random seed
; COM motion removal
; These options remove COM motion of the system
nstcomm = 10
comm-mode = Linear
comm-grps = System define = -DPOSRES: タンパク質全体に位置拘束を適用します。これにより、平衡化中にタンパク質の構造が大きく崩れるのを防ぎます。integrator = md: 分子動力学シミュレーションを行います。nsteps = 50000: 100 ps(0.002 ps/ステップ * 50000ステップ)のシミュレーション時間。tcoupl = Berendsen,pcoupl = Berendsen: 温度と圧力を目標値に調整するためのカップリングアルゴリズム。Berendsenは初期の平衡化に適しています。ref_t = 310,ref_p = 1.0: 目標温度310 K(ケルビン)、目標圧力1.0 bar(バール)。gen_vel = yes: 初期速度をランダムに生成します。refcoord_scaling = com: 圧力カップリングの際に、参照座標も重心に合わせてスケーリングします。
このMDPファイルを使って、平衡化のための実行ファイル(.tpr)を生成し、シミュレーションを実行します。
gmx grompp -f npt.mdp -c em.gro -p topol.top -r em.gro -o npt.tpr -maxwarn 3
gmx mdrun -deffnm nptgmx grompp:npt.mdp, 最小化後の構造em.gro,topol.topを基にnpt.tprを生成します。r em.groは、位置拘束の参照構造としてem.groを使用することを示します。gmx mdrun:npt.tprを使ってNPT平衡化を実行します。deffnm npt: 出力ファイルにnptというプレフィックスを付けます。
Generating Configurations
アンブレラサンプリングでは、反応座標に沿って複数の「窓」を設定し、それぞれの窓で独立したシミュレーションを行います。これらの窓の初期構造は、通常、強制的に分子を引き離す 「引き抜きシミュレーション(Steered MD or Pulling Simulation)」 によって生成されます。
Steered MD or Pulling Simulationの設定ファイル md_pull.mdp を作成します。
title = Umbrella pulling simulation
define = -DPOSRES_B
; Run parameters
integrator = md
dt = 0.002
tinit = 0
nsteps = 250000 ; 500 ps
nstcomm = 10
; Output parameters
nstxout = 5000 ; every 10 ps
nstvout = 5000
nstfout = 500
nstxtcout = 500 ; every 1 ps
nstenergy = 500
; Bond parameters
constraint_algorithm = lincs
constraints = all-bonds
continuation = yes ; continuing from NPT
; Single-range cutoff scheme
cutoff-scheme = Verlet
nstlist = 20
ns_type = grid
rlist = 1.4
rcoulomb = 1.4
rvdw = 1.4
; PME electrostatics parameters
coulombtype = PME
fourierspacing = 0.12
fourier_nx = 0
fourier_ny = 0
fourier_nz = 0
pme_order = 4
ewald_rtol = 1e-5
optimize_fft = yes
; Berendsen temperature coupling is on in two groups
Tcoupl = Nose-Hoover
tc_grps = Protein Non-Protein
tau_t = 1.0 1.0
ref_t = 310 310
; Pressure coupling is on
Pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 1.0
compressibility = 4.5e-5
ref_p = 1.0
refcoord_scaling = com
; Generate velocities is off
gen_vel = no
; Periodic boundary conditions are on in all directions
pbc = xyz
; Long-range dispersion correction
DispCorr = EnerPres
; Pull code
pull = yes
pull_ncoords = 1 ; only one reaction coordinate
pull_ngroups = 2 ; two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; harmonic potential
pull_coord1_geometry = distance ; simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; define initial COM distance > 0
pull_coord1_rate = 0.01 ; 0.01 nm per ps = 10 nm per ns
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2このMDPファイルには、アンブレラサンプリングのための「引き抜きコード(pull code)」が設定されています。
define = -DPOSRES_B: これにより、前述のtopol_Protein_chain_B.itpで追加した位置拘束が有効になり、チェーンBのみが固定されます。pull = yes: プルコードを有効にします。pull_ncoords = 1: 1つの反応座標を定義します。pull_ngroups = 2: 2つのグループ(Chain_A と Chain_B)間の距離を反応座標とします。pull_group1_name = Chain_A,pull_group2_name = Chain_B: 引き離す対象のグループの名前を指定します。pull_coord1_type = umbrella: アンブレラポテンシャルを使用しますが、この段階では「引き抜き」として機能させます。pull_coord1_geometry = distance: 距離を反応座標とします。pull_coord1_dim = N N Y: Z軸方向(Yが3番目の次元を表す)にのみ引き抜きを行います。X, Y方向は固定します。pull_coord1_rate = 0.01: 引き抜き速度を0.01 nm/ps(10 nm/ns)に設定します。pull_coord1_k = 1000: 引き抜きに使う力の定数。
引き抜きを行うには、Chain_A と Chain_B というグループを定義する必要があります。gmx make_ndx コマンドを使ってインデックスファイルを作成します。以下を実行してください。
gmx make_ndx -f npt.gro -n index.ndxコマンドを実行すると、対話形式でグループを定義するように求められます。
:-) GROMACS - gmx make_ndx, 2024 (-:
Executable: /usr/local/gromacs/bin/gmx
Data prefix: /usr/local/gromacs
Working dir: /home/shizuku/umbrella_sampling
Command line: gmx make_ndx -f npt.gro -n index.ndx
Reading structure file
Going to read 1 old index file(s) 0 System : 33207 atoms 1 Protein : 1130 atoms 2 Protein-H : 920 atoms 3 C-alpha : 135 atoms 4 Backbone : 400 atoms 5 MainChain : 540 atoms 6 MainChain+Cb : 645 atoms 7 MainChain+H : 670 atoms 8 SideChain : 460 atoms 9 SideChain-H : 380 atoms 10 Prot-Masses : 1130 atoms 11 non-Protein : 32077 atoms 12 Water : 32025 atoms 13 SOL : 32025 atoms 14 non-Water : 1182 atoms 15 Ion : 52 atoms 16 Water_and_ions : 32077 atoms nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups 'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues 't': atom type '|': or 'keep' nr 'splitat' nr 'h': help 'r': residue 'res' nr 'chain' char "name": group 'case': case sensitive 'q': save and quit 'ri': residue index > r 1-27 > name 17 Chain_A > r 28-54 > name 18 Chain_B > qr 1-27: Residue (残基) 番号1から27までを選択します。このタンパク質の構造から、これがチェーンAに対応すると仮定します。name 17 Chain_A: 選択したグループ(デフォルトで17番目のグループになる)にChain_Aという名前を付けます。r 28-54: Residue (残基) 番号28から54までを選択します。これがチェーンBに対応すると仮定します。name 28 Chain_B: 選択したグループにChain_Bという名前を付けます。q: 保存して終了します。
これにより、index.ndx ファイルが生成され、Chain_A と Chain_B というカスタムグループが定義されます。
md_pull.mdp と index.ndx を使って、引き抜きシミュレーションを実行します。
gmx grompp -f md_pull.mdp -c npt.gro -p topol.top -r npt.gro -n index.ndx -t npt.cpt -o pull.tpr -maxwarn 2
gmx mdrun -deffnm pull -pf pullf.xvg -px pullx.xvggmx grompp:md_pull.mdp, 平衡化後の構造npt.gro,topol.top,index.ndxを基にpull.tprを生成します。t npt.cptは、平衡化シミュレーションのコンティニュエーションファイル(前回のシミュレーションの状態を保存したファイル)を読み込みます。gmx mdrun:pull.tprを使って引き抜きシミュレーションを実行します。deffnm pull: 出力ファイルにpullというプレフィックスを付けます。pf pullf.xvg: 引き抜き中の力(force)をpullf.xvgに出力します。px pullx.xvg: 引き抜き中の距離(position)をpullx.xvgに出力します。
出力したpull.groとpull.xtcファイルをpymolなどで読み込むと以下のように一つの鎖が離れているのがわかります。以下はPymol-open-sourceを用いて、simulationしています。
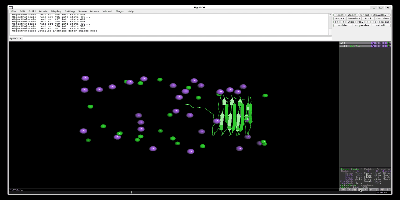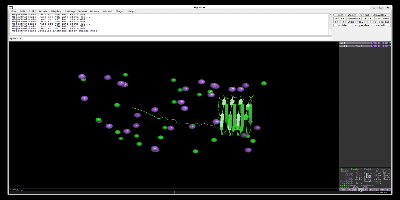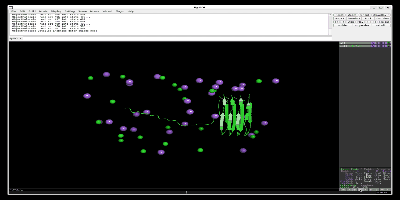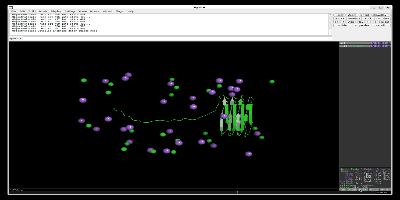
次に
引き抜きシミュレーションが完了したら、pull.xtc ファイル(トラジェクトリファイル)から各フレームでのChain_AとChain_Bの間の重心間距離を抽出し、アンブレラサンプリングの窓(初期構造)を決定します。
以下をターミナルで実行します。以下のスクリプトは以下からダウンロードしたものになります。
#!/bin/bash
#################################################
# get_distances.sh
#
# Script iteratively calls gmx distance and
# assembles a series of COM distance values
# indexed by frame number, for use in
# preparing umbrella sampling windows.
#
# Written by: Justin A. Lemkul, Ph.D.
# Contact: jalemkul@vt.edu
#
#################################################
echo 0 | gmx trjconv -s pull.tpr -f pull.xtc -o conf.gro -sep
# compute distances
for (( i=0; i<501; i++ ))
do gmx distance -s pull.tpr -f conf${i}.gro -n index.ndx -select 'com of group "Chain_A" plus com of group "Chain_B"' -oall dist${i}.xvg
done
# compile summary
touch summary_distances.dat
for (( i=0; i<501; i++ ))
do d=`tail -n 1 dist${i}.xvg | awk '{print $2}'` echo "${i} ${d}" >> summary_distances.dat rm dist${i}.xvg
done
exit;このスクリプトは以下の処理を行います。
gmx trjconv -s pull.tpr -f pull.xtc -o conf.gro -sep: 引き抜きシミュレーションの全フレームを個別の構造ファイルconf0.gro,conf1.gro, … として保存します。これにより、各フレームの構造を独立して参照できるようになります。echo 0は、Systemグループを選択するために必要です。for (( i=0; i<501; i++ )) do ... done: 各フレームについてループを実行します。gmx distance -s pull.tpr -f conf${i}.gro -n index.ndx -select 'com of group "Chain_A" plus com of group "Chain_B"' -oall dist${i}.xvg: 各conf${i}.groファイルからChain_AとChain_Bの重心間の距離を計算し、dist${i}.xvgに出力します。
for (( i=0; i<501; i++ )) do ... done: 各dist${i}.xvgファイルから距離データを抽出し、summary_distances.datにまとめます。d=tail -n 1 dist${i}.xvg | awk ‘{print $2}’:dist${i}.xvg` ファイルの最後の行から2列目(距離の値)を抽出します。echo "${i} ${d}" >> summary_distances.dat: フレーム番号と抽出した距離をsummary_distances.datに追記します。rm dist${i}.xvg: 一時的に作成したdist${i}.xvgファイルを削除します。
このスクリプトの実行後、以下のgroファイルやsummary_distances.dat ファイルが生成します。

summary_distances.dat が生成されており、以下のようになっていると思います。各フレームにおけるChain_AとChain_Bの重心間距離のリストが記録されます。
0 0.499
1 0.503
2 0.496
3 0.496
~(略)~
496 5.433
497 5.468
498 5.495
499 5.484
500 5.464この距離情報を使って、アンブレラサンプリングの窓(開始点)を決定します。例えば、距離が0.1 nm刻みで変化する窓を設定する場合、summary_distances.dat から目的の距離に近いフレームの構造ファイル confX.gro を選択することになります。
Umbrella Sampling
アンブレラサンプリングでは、反応座標(この例ではタンパク質Chain_AとChain_Bの重心間距離)に沿って複数の「窓(ウィンドウ)」を設定し、それぞれの窓で独立したシミュレーションを行います。これにより、通常の分子動力学シミュレーションではアクセスしにくい自由エネルギー障壁の領域も効率的にサンプリングできます。
一つ一つは以下の流れで行います。(ここは最後にまとめてやるので、実行しなくてよいです)
窓の初期設定とNPT平衡化
まず、引き抜きシミュレーション(Steered MD)で得られた軌道から、サンプリング窓の初期構造 を選びます。この例では、Z軸方向に0.5 nmから5.0 nmまで、約0.2 nm間隔で合計23個の窓を設定するとのことです。引き抜きシミュレーションの出力(conf*.groファイル)の中から、それぞれの窓の距離に近い構造を特定して使用します。
各窓で本格的なアンブレラサンプリングを開始する前に、それぞれの初期構造を NPTアンサンブル(定温定圧アンサンブル)で短時間平衡化 します。これは、各窓の初期構造が、その窓で設定された拘束条件下で安定した状態になるようにするためです。
この平衡化には、npt_umbrella.mdp ファイルを使用します。このMDPファイルは、アンブレラサンプリングの本番用MDPファイルと似ていますが、主な違いは、速度を再度ランダムに生成する (gen_vel = yes) 点です。これにより、各窓で独立した初期状態から平衡化を開始できます。
npt_umbrella.mdp (あとで使うので、作成しておいてください)
title = Umbrella pulling simulation
define = -DPOSRES_B
; Run parameters
integrator = md
dt = 0.002
tinit = 0
nsteps = 50000 ; 100 ps
nstcomm = 10
; Output parameters
nstvout = 5000 ; every 10 ps
nstfout = 5000
nstxout-compressed = 5000
nstenergy = 5000
; Bond parameters
constraint_algorithm = lincs
constraints = all-bonds
continuation = no
; Single-range cutoff scheme
cutoff-scheme = Verlet
nstlist = 20
ns_type = grid
rlist = 1.4
rcoulomb = 1.4
rvdw = 1.4
; PME electrostatics parameters
coulombtype = PME
fourierspacing = 0.12
fourier_nx = 0
fourier_ny = 0
fourier_nz = 0
pme_order = 4
ewald_rtol = 1e-5
optimize_fft = yes
; Berendsen temperature coupling is on in two groups
Tcoupl = Berendsen
tc_grps = Protein Non-Protein
tau_t = 0.5 0.5
ref_t = 310 310
; Pressure coupling is on
Pcoupl = Berendsen
pcoupltype = isotropic
tau_p = 1.0
compressibility = 4.5e-5
ref_p = 1.0
refcoord_scaling = com
; Generate velocities is on
gen_vel = yes
gen_temp = 310
; Periodic boundary conditions are on in all directions
pbc = xyz
; Long-range dispersion correction
DispCorr = EnerPres
; Pull code
pull = yes
pull_ncoords = 1 ; only one reaction coordinate
pull_ngroups = 2 ; two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; harmonic potential
pull_coord1_geometry = distance ; simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; define initial COM distance > 0
pull_coord1_rate = 0.0 ; restrain in place
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
; 新しく追加する行
pull_group1_pbcatom = 113 ; Chain_Aの中心に近い原子のインデックスを指定 (例として113)
pull-pbc-ref-prev-step-com = yesgmx grompp で入力ファイルを生成 (ここは最後にまとめてやるので、実行しなくてよいです)
平衡化のための.tprファイル(シミュレーション実行ファイル)を生成します。23個の窓それぞれについて、このコマンドを実行します。
gmx grompp -f npt_umbrella.mdp -c conf6.gro -p topol.top -r conf6.gro -n index.ndx -o npt0.tpr
# ... 途中省略 ...
gmx grompp -f npt_umbrella.mdp -c conf449.gro -p topol.top -r conf449.gro -n index.ndx -o npt22.tprf npt_umbrella.mdp: NPT平衡化のための設定ファイル。c confX.gro: 各窓の初期構造ファイル。conf6.groからconf449.groまで、それぞれの窓の開始距離に対応する構造を使用します。p topol.top: メイントポロジーファイル。r confX.gro: 位置拘束の参照構造としても、その窓の初期構造を使用します。これにより、初期平衡化中に構造が大きく崩れるのを防ぎます。n index.ndx: 以前に作成した、Chain_AとChain_Bのグループ定義を含むインデックスファイル。o nptX.tpr: 出力される.tprファイル。各窓で異なる名前(npt0.tprからnpt22.tpr)を付けます。
gmx mdrun で平衡化シミュレーションを実行(ここは最後にまとめてやるので、実行しなくてよいです)
生成した.tprファイルを使って、各窓のNPT平衡化シミュレーションを実行します。
gmx mdrun -deffnm npt0
# ... 途中省略 ...
gmx mdrun -deffnm npt22deffnm nptX: 出力ファイル(ログ、構造、エネルギーなど)にnptXというプレフィックスを付けます。これにより、各窓の出力が区別されます。
アンブレラサンプリング本番シミュレーション(ここは最後にまとめてやるので、実行しなくてよいです)
各窓でNPT平衡化が完了したら、いよいよ本番のアンブレラサンプリング を開始します。このフェーズでは、各窓の反応座標値を中心に、調和ポテンシャルを用いて分子を拘束しながら、より長時間のシミュレーションを行います。
この本番シミュレーションには、md_umbrella.mdp ファイルを使用します。
md_umbrella.mdp**(あとで使うので、作成しておいてください)**
title = Umbrella pulling simulation
define = -DPOSRES_B
; Run parameters
integrator = md
dt = 0.002
tinit = 0
nsteps = 5000000 ; 10 ns
nstcomm = 10
; Output parameters
nstxout-compressed = 5000 ; every 10 ps
nstenergy = 5000
; Bond parameters
constraint_algorithm = lincs
constraints = all-bonds
continuation = yes
; Single-range cutoff scheme
cutoff-scheme = Verlet
nstlist = 20
ns_type = grid
rlist = 1.4
rcoulomb = 1.4
rvdw = 1.4
; PME electrostatics parameters
coulombtype = PME
fourierspacing = 0.12
fourier_nx = 0
fourier_ny = 0
fourier_nz = 0
pme_order = 4
ewald_rtol = 1e-5
optimize_fft = yes
; Berendsen temperature coupling is on in two groups
Tcoupl = Nose-Hoover
tc_grps = Protein Non-Protein
tau_t = 1.0 1.0
ref_t = 310 310
; Pressure coupling is on
Pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 1.0
compressibility = 4.5e-5
ref_p = 1.0
refcoord_scaling = com
; Generate velocities is off
gen_vel = no
; Periodic boundary conditions are on in all directions
pbc = xyz
; Long-range dispersion correction
DispCorr = EnerPres
; Pull code
pull = yes
pull_ncoords = 1 ; only one reaction coordinate
pull_ngroups = 2 ; two groups defining one reaction coordinate
pull_group1_name = Chain_A
pull_group2_name = Chain_B
pull_coord1_type = umbrella ; harmonic potential
pull_coord1_geometry = distance ; simple distance increase
pull_coord1_dim = N N Y
pull_coord1_groups = 1 2
pull_coord1_start = yes ; define initial COM distance > 0
pull_coord1_rate = 0.0 ; restrain in place
pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
; 新しく追加する行
pull_group1_pbcatom = 113 ; Chain_Aの中心に近い原子のインデックスを指定 (例として113)
pull-pbc-ref-prev-step-com = yespull_coord1_rate = 0.0: 引き抜き速度がゼロ に設定されています。これが最も重要な変更点です。これにより、分子を反応座標に沿って動かすのではなく、定義された窓の空間内で拘束 することになります。pull_coord1_start = yes: これは、シミュレーション開始時のCOM(重心)間距離が、その窓の参照距離(中心) として自動的に設定されることを意味します。そのため、各窓ごとに参照距離(pull_coord1_init)を手動で定義する必要がありません。非常に便利な機能です。
本番のアンブレラサンプリングのための.tprファイルを生成します。ここでも23個の窓それぞれについてコマンドを実行します。
gmx grompp -f md_umbrella.mdp -c npt0.gro -t npt0.cpt -p topol.top -r npt0.gro -n index.ndx -o umbrella0.tpr
# ... 途中省略 ...
gmx grompp -f md_umbrella.mdp -c npt22.gro -t npt22.cpt -p topol.top -r npt22.gro -n index.ndx -o umbrella22.tprf md_umbrella.mdp: 本番のアンブレラサンプリングのための設定ファイル。c nptX.gro: 前のNPT平衡化ステップで得られた最終構造。t nptX.cpt: 前のNPT平衡化ステップのコンティニュエーションファイル。これにより、前のシミュレーションの速度や状態を引き継いで、シームレスにシミュレーションを継続できます。p topol.top,r nptX.gro,n index.ndx: これらはこれまでのステップと同様です。o umbrellaX.tpr: 出力される本番シミュレーション用の.tprファイル。各窓で異なる名前(umbrella0.tprからumbrella22.tpr)を付けます。
gmx mdrun で本番シミュレーションを実行(ここは最後にまとめてやるので、実行しなくてよいです)
生成した.tprファイルを使って、各窓の本番アンブレラサンプリングシミュレーションを実行します。これはデータ収集のフェーズであり、通常、各窓で数ナノ秒(ns)から数十nsといった長時間のシミュレーションを行います。
gmx mdrun -deffnm umbrella0
# ... 途中省略 ...
gmx mdrun -deffnm umbrella22deffnm umbrellaX: 出力ファイルにumbrellaXというプレフィックスを付けます。
まず、各アンブレラ窓の初期平衡化のための設定ファイル npt_umbrella.mdp を作成します。これは、引き抜きシミュレーションで生成された各窓の初期構造を、目的の拘束条件下で平衡化するためのものです。
(ここから実行してください!)
上記はひとつひとつ行うのは時間がかかるため、チュートリアルで、一気にできるスクリプトが提供されていますが、python 2で書かれているので、今は実行できません。python 3のスクリプトはを作成したので、コピペして、setupUmbrella_py3.py として、保存してください
setupUmbrella_py3.py
#!/usr/bin/env python
__description__ = \
"""
Take a file containing frame number vs. distance, then identify frames that
sample those distances at approximately sample_interval. Optionally takes an
arbitrary number of template files. The program searches the contents of these
files for a search string (by default XXX), replaces the search string with the
frame number, then writes out each file with a unique, frame-specific filename.
This last feature means that this script can be used to automatically generate
input files to run each umbrella sampling simulation.
v. 0.1: fixed bug where the script would choke on distance_files that went by frame intervals besides steps of 1.
"""
__author__ = "Michael J. Harms [harmsm@gmail.com]"
__date__ = "120314"
__usage__ = "setupUmbrella.py distance_file sample_interval [--output=summary_file] [template_file1 template_file2 ...]"
__version__ = "0.1"
import sys, os, re
def readDistanceFile(distance_file): """ Read a distance file that correlates file number with center-of-mass distance between pulling groups. Format is: 0 com0 1 com1 2 com2 ... If this data was generated using distances.pl (Justin Lemkul), it could have multiple com calculations in the same file. This function starts from the bottom of the file and only takes the last output. """ # Read file f = open(distance_file,'r') lines = f.readlines() f.close() # Read the data from the bottom out_dict = {} for i in range(len(lines)-1,-1,-1): # Split on white-space; grab frame/distance columns = lines[i].split() key = int(columns[0]) value = float(columns[1]) # If we've already seen this key, we're done reading the unique data # from the file. if key in out_dict: break else: out_dict[key] = value # Now put the values into a simple list, sorting to make sure that they are # in the correct order. keys = list(out_dict.keys()) # Python 3: dict.keys() returns a view, need to convert to list for .sort() keys.sort() out = [(k,out_dict[k]) for k in keys] return out
def sampleDistances(distance_table,sample_interval): """ Go through the distances list and sample frames at sample_interval. Appropriate samples are identified by looking forward through the distances to find the one that is closest to current_distance + sample_interval. """ distances = [d[1] for d in distance_table] current_index = 0 sampled_indexes = [current_index] while current_index < len(distances): target_distance = distances[current_index] + sample_interval # Walk through the rest of the distances list and find the distance # that is closest to the target distance onward = [abs(target_distance-d) for d in distances[current_index:]] next_index = onward.index(min(onward)) + current_index # If we run out of distances to compare, our next_index will be the # current index. This means we're done. Otherwise, record that we # found a new index. if current_index == next_index: break else: sampled_indexes.append(next_index) current_index = next_index return sampled_indexes
def createOutputFile(template_file,frame_number,search_string="XXX"): """ Look for instances of the search string in a template file and replace with the frame number in a new file. """ out_file = "frame-%i_%s" % (frame_number,template_file) # Prompt the user before wiping out an existing file if os.path.exists(out_file): answer = input("%s exists! Overwrite (y|n)?" % out_file) # Changed raw_input to input answer = answer[0].lower() if answer != "y": return None # Read the contents of the template file f = open(template_file,'r') file_contents = f.read() f.close() # Write out the template file contents, replacing all instances of # the search string with the frame number f = open(out_file,'w') f.write(re.sub(search_string,"%i" % frame_number,file_contents)) f.close()
def main(argv=None, output_summary_file=None): # output_summary_file パラメータを追加 """ Parse command line, etc. """ if argv == None: argv = sys.argv[1:] # Parse required command line arguments try: distance_file = argv[0] sample_interval = float(argv[1]) # template_files は argv から自動的に続く引数として取得 template_files = argv[2:] except (IndexError,ValueError): err = "Incorrect command line arguments!\n\n%s\n\n" % __usage__ raise IOError(err) # See if a template file has been specified # template_files が空リストでないかどうかのチェックはそのまま # template_files = argv[2:] # Figure out which frames to use distance_table = readDistanceFile(distance_file) sampled_indexes = sampleDistances(distance_table,sample_interval) # If any template files were specified, use them to make frame-specific # output if len(template_files) != 0: print("Creating frame-specific output for files:") print("\n".join(template_files)) for t in template_files: for i in sampled_indexes: frame = distance_table[i][0] createOutputFile(t,frame,search_string="XXX") # Print out summary of the frames we identified out = ["%10s%10s%10s\n" % ("frame","dist","d_dist")] for i in range(len(sampled_indexes)): frame = distance_table[sampled_indexes[i]][0] dist = distance_table[sampled_indexes[i]][1] if i == 0: delta_dist = "%10s" % "NA" else: prev_dist = distance_table[sampled_indexes[i-1]][1] delta_dist = "%10.3f" % (dist - prev_dist) out.append("%10i%10.3f%s\n" % (frame,dist,delta_dist)) summary_output = "".join(out) # 変更点: output_summary_file が指定されていればファイルに書き込み、そうでなければ標準出力 if output_summary_file: try: with open(output_summary_file, 'w') as f: f.write(summary_output) print(f"Summary written to {output_summary_file}") # このメッセージは標準出力に出る except IOError as e: print(f"Error writing summary to file {output_summary_file}: {e}") print(summary_output) # ファイル書き込み失敗時は標準出力にフォールバック else: print(summary_output) # 標準出力に書き出す return out # 戻り値は以前と同じリストだが、直接出力には使われない
# If called from the command line...
if __name__ == "__main__": parsed_output_file = None processed_argv = [] # main 関数に渡す引数を格納するリスト # sys.argv から --output=filename 引数を抽出し、残りを processed_argv に格納 i = 0 # sys.argv[0] はスクリプト名自身なので、sys.argv[1:] から開始 while i < len(sys.argv[1:]): arg = sys.argv[1+i] if arg.startswith("--output="): parsed_output_file = arg.split("=")[1] else: processed_argv.append(arg) i += 1 # main 関数を呼び出し、処理済み引数と出力ファイル名を渡す main(argv=processed_argv, output_summary_file=parsed_output_file)このPythonスクリプト setupUmbrella.py は、分子動力学シミュレーションのアンブレラサンプリングを行う際に、多数のシミュレーション窓の初期設定を自動化するためのものです。
簡単に言うと、以下のような流れで動作します。
- 距離データの読み込み: まず、引き抜きシミュレーション(Steered MD)の結果として得られた、シミュレーションの各時点(フレーム)における分子間の距離データが書かれたファイル(例:
summary_distances.dat)を読み込みます。 - サンプリング窓の特定: 次に、読み込んだ距離データから、ユーザーが指定した間隔(例: 0.2 nm)で均等にサンプリングされるように、アンブレラサンプリングの「窓」として使用すべきフレーム番号を特定します。これは、目標とする距離に最も近いフレームを探すことで行われます。
- 入力ファイルの自動生成: 最後に、特定された各フレーム番号(窓)に対して、提供されたテンプレートファイル(通常はGROMACSの
.mdpファイルなど)を基に、新しい入力ファイルを自動で生成します。この際、テンプレートファイル内の特定のプレースホルダー文字列(デフォルトはXXX)を、対応するフレーム番号に置き換えます。
このスクリプトを使うことで、手動で数十〜数百もの入力ファイルを作成する手間を省き、アンブレラサンプリングのセットアッププロセスを大幅に効率化できます。
以下を実行してください。
python setupUmbrella_py3.py summary_distances.dat 0.2 --output=umbrella_summary.txt npt_umbrella.mdp md_umbrella.mdpsummary_distances.datを読み込み、そこから約0.2 nm間隔で距離が変化するシミュレーションフレームを特定します。- 特定された各フレームに対して、
npt_umbrella.mdpとmd_umbrella.mdpのテンプレートを基に、新しい.mdpファイルを生成します。これらの新しいファイル名や内容には、対応するフレーム番号が埋め込まれます。
これにより、数十個にもなる可能性のあるアンブレラサンプリングの各窓のシミュレーション設定ファイル(.mdp)が自動で準備され、その後のGROMACSでのシミュレーション実行が効率化されます。
(ミスってファイルを作成した場合、消したいときは以下を実行してください。)
rm -f frame-*_npt_umbrella.mdp frame-*_md_umbrella.mdpまずは上記のNPT平衡化をすべてのものに対して行います。
以下をコピーし、run_npt_equilibration_loop.pyを作成してください。
run_npt_equilibration_loop.py
import subprocess
import os
import argparse
def run_gromacs_command(command, dry_run=False): """ GROMACSコマンドを実行するヘルパー関数。 dry_run=Trueの場合、コマンドを表示するだけで実行しない。 """ command_str = ' '.join(command) print(f"\nExecuting: {command_str}") if not dry_run: try: # check=True: コマンドがエラーコードで終了した場合、CalledProcessErrorを発生させる # capture_output=True=False: 標準出力とエラー出力をリアルタイムで表示するためにFalseにする # text=True: stdoutとstderrをテキストとしてデコードする # Popen を使用してリアルタイム出力を実現 process = subprocess.Popen(command, stdout=subprocess.PIPE, stderr=subprocess.STDOUT, text=True, bufsize=1) for line in process.stdout: print(line, end='') # 行ごとに表示 process.wait() # プロセスが終了するのを待つ if process.returncode != 0: print(f"Error executing command: {command_str}") print(f"Return Code: {process.returncode}") # stdout/stderrはすでに表示されているので、ここでは追加で表示しない exit(1) # エラーが発生したらスクリプトを終了 except FileNotFoundError: print(f"Error: GROMACS command '{command[0]}' not found. Make sure GROMACS is in your PATH.") exit(1)
def parse_summary_file(summary_file_path): """ setupUmbrella.py が出力したサマリーファイルからフレーム番号のリストをパースします。 """ frames = [] if not os.path.exists(summary_file_path): print(f"Error: Summary file '{summary_file_path}' not found. Please ensure setupUmbrella.py was run and outputted to this file.") exit(1) print(f"--- Parsing initial configuration frames from {summary_file_path} ---") with open(summary_file_path, 'r') as f: # "frame dist d_dist" の行より後の行を処理 start_parsing = False for line in f: if line.strip().startswith("frame dist d_dist"): start_parsing = True continue if start_parsing: # 空行やコメント行をスキップ if not line.strip() or line.strip().startswith("#"): continue columns = line.split() try: frame_num = int(columns[0]) frames.append(frame_num) except (ValueError, IndexError): # 数値に変換できない行はスキップ(例:区切り線など) continue if not frames: print("Error: No initial configuration frames found in the summary file. Please check the file content.") exit(1) print(f"Identified {len(frames)} windows with frames: {frames}") return frames
def main(): parser = argparse.ArgumentParser(description="Run NPT equilibration for each window based on setupUmbrella.py output.") parser.add_argument("--summary", type=str, default="umbrella_summary.txt", help="Path to the summary file generated by setupUmbrella.py (default: umbrella_summary.txt)") parser.add_argument("--npt_mdp", type=str, default="npt_umbrella.mdp", help="NPT equilibration MDP template file (default: npt_umbrella.mdp)") parser.add_argument("--topol", type=str, default="topol.top", help="Main topology file (default: topol.top)") parser.add_argument("--index", type=str, default="index.ndx", help="Index file (default: index.ndx)") parser.add_argument("--maxwarn", type=int, default=5, help="Maximum number of warnings tolerated by grompp before exiting (default: 5).") parser.add_argument("--dry-run", action="store_true", help="If set, commands will be printed but not executed.") args = parser.parse_args() # setupUmbrella.py のサマリーファイルからフレーム番号を読み込む initial_conf_frames = parse_summary_file(args.summary) num_windows = len(initial_conf_frames) print("\n" + "="*50) print("--- Running NPT Equilibration for each window ---") print("="*50) for i in range(num_windows): conf_frame_num = initial_conf_frames[i] conf_gro_file = f"conf{conf_frame_num}.gro" output_tpr_file = f"npt{i}.tpr" output_deffnm = f"npt{i}" # 1. grompp grompp_command = [ "gmx", "grompp", "-f", args.npt_mdp, "-c", conf_gro_file, "-p", args.topol, "-r", conf_gro_file, "-n", args.index, "-o", output_tpr_file, "-maxwarn", str(args.maxwarn) ] run_gromacs_command(grompp_command, args.dry_run) # 2. mdrun mdrun_command = [ "gmx", "mdrun", "-deffnm", output_deffnm, "-v" # 進行状況を表示 ] run_gromacs_command(mdrun_command, args.dry_run) print("\n" + "="*50) print("--- NPT Equilibration for all windows complete ---") print("="*50) if args.dry_run: print("Note: This was a DRY RUN. No commands were actually executed.")
if __name__ == "__main__": main()以下で実行してください。
python run_npt_equilibration_loop.pyrun_npt_equilibration_loop.py の簡単な流れ
- 必要な情報の読み込み:
- まず、以前に実行した
setupUmbrella.pyが作成した、アンブレラサンプリングの「窓」となる各フレームの番号が書かれたファイル(例:umbrella_summary.txt)を読み込みます。 - また、NPT平衡化に必要なGROMACSの設定ファイル名(
.mdp、トポロジー、インデックスファイルなど)も引数から受け取ります。
- まず、以前に実行した
- 窓ごとの繰り返し処理:
- 読み込んだフレーム番号の数だけ(つまり、設定された窓の数だけ)処理を繰り返します。
- 各窓でのシミュレーション準備 (grompp):
- 現在の窓に対応する
conf*.groファイル(特定された初期構造)と、NPT平衡化の設定ファイル(npt_umbrella.mdp)を使って、gmx gromppコマンドを実行します。 - これにより、その窓のNPT平衡化シミュレーションを実行するためのバイナリファイル(
nptX.tpr)が作成されます。
- 現在の窓に対応する
- 各窓でのシミュレーション実行 (mdrun):
- 作成された
nptX.tprファイルを使って、gmx mdrunコマンドを実行し、NPT平衡化シミュレーションを開始します。 - この際、
vオプションにより、シミュレーションの進捗状況(残り時間など)がコンソールにリアルタイムで表示されます。
- 作成された
- エラーチェックと終了:
- 各GROMACSコマンドの実行後、エラーがないかを確認します。もしエラーが発生した場合は、その時点でスクリプトを停止し、エラーメッセージを表示します。
- 全ての窓のNPT平衡化が成功すれば、スクリプトは完了メッセージを表示して終了します。
まとめると、このスクリプトは、setupUmbrella.pyで選ばれた初期構造のリストに基づいて、それぞれの構造に対して「シミュレーションの準備」と「短時間の安定化シミュレーション」を順番に自動で行うツールです。
同様にumbrella simulationもすべてにおいて行っていきます。
run_umbrella_data_collection_loop.py
import subprocess
import os
import argparse
def run_gromacs_command(command, dry_run=False): """ GROMACSコマンドを実行するヘルパー関数。 dry_run=Trueの場合、コマンドを表示するだけで実行しない。 """ command_str = ' '.join(command) print(f"\nExecuting: {command_str}") if not dry_run: try: process = subprocess.Popen(command, stdout=subprocess.PIPE, stderr=subprocess.STDOUT, text=True, bufsize=1) for line in process.stdout: print(line, end='') process.wait() if process.returncode != 0: print(f"Error executing command: {command_str}") print(f"Return Code: {process.returncode}") exit(1) except FileNotFoundError: print(f"Error: GROMACS command '{command[0]}' not found. Make sure GROMACS is in your PATH.") exit(1)
def parse_summary_file(summary_file_path): """ setupUmbrella.py が出力したサマリーファイルからフレーム番号のリストをパースします。 アンブレラ窓の数を決定するために使用します。 """ frames = [] if not os.path.exists(summary_file_path): print(f"Error: Summary file '{summary_file_path}' not found. Please ensure setupUmbrella.py was run and outputted to this file.") exit(1) print(f"--- Parsing initial configuration frames from {summary_file_path} ---") with open(summary_file_path, 'r') as f: start_parsing = False for line in f: if line.strip().startswith("frame dist d_dist"): start_parsing = True continue if start_parsing: if not line.strip() or line.strip().startswith("#"): continue columns = line.split() try: frame_num = int(columns[0]) frames.append(frame_num) except (ValueError, IndexError): continue if not frames: print("Error: No initial configuration frames found in the summary file. Please check the file content.") exit(1) print(f"Identified {len(frames)} windows with frames: {frames}") return frames
def main(): parser = argparse.ArgumentParser(description="Run Umbrella Sampling data collection for each window.") parser.add_argument("--summary", type=str, default="umbrella_summary.txt", help="Path to the summary file generated by setupUmbrella.py (default: umbrella_summary.txt)") parser.add_argument("--md_umbrella_mdp", type=str, default="md_umbrella.mdp", help="Umbrella sampling MDP template file (default: md_umbrella.mdp)") parser.add_argument("--topol", type=str, default="topol.top", help="Main topology file (default: topol.top)") parser.add_argument("--index", type=str, default="index.ndx", help="Index file (default: index.ndx)") parser.add_argument("--maxwarn", type=int, default=5, help="Maximum number of warnings tolerated by grompp before exiting (default: 5).") parser.add_argument("--dry-run", action="store_true", help="If set, commands will be printed but not executed.") args = parser.parse_args() # setupUmbrella.py のサマリーファイルから窓の数を読み込む # ここで取得するフレーム番号自体は直接使わないが、窓の数を決定するために必要 initial_conf_frames = parse_summary_file(args.summary) num_windows = len(initial_conf_frames) print("\n" + "="*50) print("--- Running Umbrella Sampling Data Collection for each window ---") print("="*50) for i in range(num_windows): # NPT平衡化ステップで生成されたファイルを基にする npt_equil_gro_file = f"npt{i}.gro" npt_equil_cpt_file = f"npt{i}.cpt" umbrella_output_tpr = f"umbrella{i}.tpr" umbrella_output_deffnm = f"umbrella{i}" # 1. grompp for Umbrella Sampling umbrella_grompp_command = [ "gmx", "grompp", "-f", args.md_umbrella_mdp, "-c", npt_equil_gro_file, # NPT平衡化の最終構造 "-t", npt_equil_cpt_file, # NPT平衡化のコンティニュエーションファイル "-p", args.topol, "-r", npt_equil_gro_file, # -r オプションは拘束の参照構造 (通常-cと同じ) "-n", args.index, "-o", umbrella_output_tpr, "-maxwarn", str(args.maxwarn) ] run_gromacs_command(umbrella_grompp_command, args.dry_run) # 2. mdrun for Umbrella Sampling umbrella_mdrun_command = [ "gmx", "mdrun", "-deffnm", umbrella_output_deffnm, "-v" # 進捗状況を表示 ] run_gromacs_command(umbrella_mdrun_command, args.dry_run) print("\n" + "="*50) print("--- Umbrella Sampling Data Collection for all windows complete ---") print("="*50) if args.dry_run: print("Note: This was a DRY RUN. No commands were actually executed.")
if __name__ == "__main__": main()以下で実行してください。
python run_umbrella_data_collection_loop.pyrun_umbrella_data_collection_loop.py の簡単な流れ
- 必要な情報の読み込み:
- まず、
setupUmbrella.pyが作成した、アンブレラ窓のフレーム番号が書かれたサマリーファイル(umbrella_summary.txt)を読み込み、実行すべき窓の数を把握します。 - アンブレラサンプリング本番に必要なGROMACSの設定ファイル名(
md_umbrella.mdp、トポロジー、インデックスファイルなど)も引数から受け取ります。
- まず、
- 窓ごとの繰り返し処理:
- 把握した窓の数だけ(例:
0から22まで)処理を繰り返します。
- 把握した窓の数だけ(例:
- 各窓でのシミュレーション準備 (grompp):
- 現在の窓に対応する、NPT平衡化で得られた最終構造ファイル(
nptX.gro)と、その時点の情報を引き継ぐためのチェックポイントファイル(nptX.cpt)を入力として使用します。 - これらのファイルと、アンブレラサンプリング本番の設定ファイル(
md_umbrella.mdp)を使って、gmx gromppコマンドを実行します。 - これにより、その窓のアンブレラサンプリングを実行するためのバイナリファイル(
umbrellaX.tpr)が作成されます。
- 現在の窓に対応する、NPT平衡化で得られた最終構造ファイル(
- 各窓でのシミュレーション実行 (mdrun):
- 作成された
umbrellaX.tprファイルを使って、gmx mdrunコマンドを実行し、アンブレラサンプリングの本番データ収集シミュレーションを開始します。 deffnm umbrellaXにより、シミュレーションの結果ファイル(トラジェクトリやログなど)がumbrellaX.*という名前で保存されます。vオプションにより、シミュレーションの進捗状況(残り時間など)がコンソールにリアルタイムで表示されます。
- 作成された
- エラーチェックと終了:
- 各GROMACSコマンドの実行後、エラーがないかを確認します。もしエラーが発生した場合は、その時点でスクリプトを停止し、問題があったことを報告します。
- 全ての窓のデータ収集シミュレーションが成功すれば、スクリプトは完了メッセージを表示して終了します。
1日ほどかかり、umbrella1-25の色々なファイルが生成されます。

Data Analysis (データ解析)
このステップでは、アンブレラサンプリングシミュレーションで集めたデータを使って、最終的な目標であるPMF(Potential of Mean Force:平均力ポテンシャル)を計算する方法を学びます。PMFを計算することで、結合や結合解除(バインディング/アンバインディング)といったプロセスにかかるΔG(自由エネルギー変化)**が分かります。
まず以下をproduction_dat_files.py という形で保存してください。
import os
def parse_summary_file_for_num_windows(summary_file_path="umbrella_summary.txt"): """ setupUmbrella.py が出力したサマリーファイルから窓の数を決定します。 """ frames = [] if not os.path.exists(summary_file_path): print(f"Error: Summary file '{summary_file_path}' not found. Please ensure setupUmbrella.py was run and outputted to this file.") return None # エラーの場合はNoneを返す with open(summary_file_path, 'r') as f: start_parsing = False for line in f: if line.strip().startswith("frame dist d_dist"): start_parsing = True continue if start_parsing: if not line.strip() or line.strip().startswith("#"): continue columns = line.split() try: frame_num = int(columns[0]) frames.append(frame_num) except (ValueError, IndexError): continue if not frames: print("Error: No initial configuration frames found in the summary file. Cannot determine number of windows.") return None num_windows = len(frames) print(f"Identified {num_windows} windows from the summary file.") return num_windows
# --- ここからが.datファイル作成の主要部分 ---
if __name__ == "__main__": # サマリーファイルのパスを適宜変更してください summary_file = "umbrella_summary.txt" # WHAM解析に使用するpullファイルのタイプを選択 ('pullx' または 'pullf') # チュートリアルでは 'pullf' を使用します。 pull_file_type = "pullf" num_windows = parse_summary_file_for_num_windows(summary_file) if num_windows is None: print("Failed to determine number of windows. Exiting.") exit(1) print("\n" + "="*50) print("--- Creating .dat files for WHAM analysis ---") print("="*50) tpr_files_dat = "tpr-files.dat" pull_files_dat = f"{pull_file_type}-files.dat" # 1. tpr-files.dat の作成 print(f"Creating {tpr_files_dat}...") with open(tpr_files_dat, "w") as f: for i in range(num_windows): f.write(f"umbrella{i}.tpr\n") print(f" {tpr_files_dat} created with {num_windows} entries.") # 2. pullX-files.dat (または pullF-files.dat) の作成 print(f"Creating {pull_files_dat}...") with open(pull_files_dat, "w") as f: for i in range(num_windows): f.write(f"umbrella{i}_{pull_file_type}.xvg\n") print(f" {pull_files_dat} created with {num_windows} entries.") print("\n--- .dat file creation complete ---") print(f"You can now use these files ({tpr_files_dat} and {pull_files_dat}) with gmx wham.")以下で実行します。
python production_dat_files.pyproduction_dat_files.py の簡単な流れ
このスクリプトは、アンブレラサンプリングによって得られた複数のシミュレーションデータから、WHAM解析の入力として必要なファイルリスト(.datファイル)を自動的に生成します。
- 必要な情報の読み込み:
- まず、
setupUmbrella.pyスクリプトが生成したサマリーファイル(デフォルトではumbrella_summary.txt)を読み込み、アンブレラサンプリングで実行された窓の総数を特定します。この窓の数は、WHAM解析のために生成するファイルの数を決定するために不可欠です。 - また、WHAM解析に使用するGROMACSの
pullファイルのタイプ(pullxまたはpullf)を選択できます。チュートリアルでは通常pullfが使用されています。
- まず、
.datファイルの生成:- 特定された窓の数に基づいて、WHAM解析に必要な2種類の
.datファイルを生成します。tpr-files.dat: このファイルには、各アンブレラ窓のGROMACS実行ファイル(.tprファイル)へのパスが一覧で記述されます。WHAMはこれらの.tprファイルから各窓のバイアス情報を読み取ります。pullX-files.dat(またはpullF-files.dat): このファイルには、各アンブレラ窓から得られたバイアス座標(pullx.xvg)またはバイアス力(pullf.xvg)のデータファイルへのパスが一覧で記述されます。WHAMはこれらのファイルから、各窓での系の位置または力の情報を取得し、ポテンシャル平均力を計算します。
- 特定された窓の数に基づいて、WHAM解析に必要な2種類の
- エラーチェックと終了:
- サマリーファイルが見つからない場合や、窓の数を特定できなかった場合は、適切なエラーメッセージを表示してスクリプトは終了します。
- 全ての
.datファイルが正常に作成されると、完了メッセージが表示され、WHAM解析に進む準備ができたことがユーザーに伝えられます。
このスクリプトを実行することで、手動で.datファイルを作成する手間を省き、GROMACSのgmx whamコマンドをすぐに実行できる状態にすることができます。
続いて、以下を実行してください。
gmx wham -it tpr-files.dat -if pullf-files.dat -o pmf -hist -unit kCalgmx wham は、GROMACSというソフトで使われるコマンドで、アンブレラサンプリングというシミュレーション結果を分析するためのものです。この分析でPMF(平均力ポテンシャル)という、ある現象が起こるために必要なエネルギーの変化を計算します。
このコマンドは、簡単に言うと以下のようなことをgmx whamに指示しています。
gmx wham: WHAM解析を始めます。it tpr-files.dat: シミュレーションの設定ファイル(.tprファイル)がリストされたファイルを見てね。これで、どんな「傘」(バイアス)がかかっていたかが分かります。if pullf-files.dat: 各シミュレーションで測定された「力」のデータ(pullf.xvgファイル)がリストされたファイルを見てね。この力を使ってPMFを計算します。o pmf: 計算結果のPMFをpmf.xvgという名前のファイルに保存します。hist: おまけで、各シミュレーションのデータの分布(ヒストグラム)も出力します。unit kCal: エネルギーの単位は「キロカロリー/モル」で表示します。
つまり、このコマンドは「アンブレラシミュレーションのデータを使って、対象の現象が起こるのにどれくらいのエネルギーが必要か(PMF)を計算し、その結果をファイルに保存する」という意味になります。
続いて、以下でpmfのグラフを作成します。
xmgrace pmf.xvg
グラフ「Umbrella potential」の解説
このグラフは、特定の現象が起こるための「エネルギーの坂道」を示しています。
- X軸 (ξ (nm)):
- 何かの「変化の度合い」や「距離」を表します。単位はnm(ナノメートル)。
- 右に行くほど、その変化が進んだり、距離が離れたりします。
- Y軸 (E (kcal mol⁻¹)):
- その変化に必要な「自由エネルギー(エネルギーの高さ)」を表します。単位はkcal/mol(キロカロリー/モル)。
- 上にいくほど、その状態になるのが難しく(エネルギーが必要に)なります。
グラフの解釈
- グラフは、X軸が約0.5nmから3.7nmまで進むにつれて、エネルギーが約0から48 kcal/molまで急激に上昇しています。
- これは、この変化を起こすには、かなりのエネルギーが必要であることを示しています。まるで、高い坂道を登るようなものです。
- X軸が約3.7nmから5.5nmになると、エネルギーはほぼ横ばい(約48 kcal/mol)になります。
- これは、もうこれ以上変化してもエネルギーはほとんど変わらない、つまり「ゴールに達した」ような状態を示しています。
簡単に言うと、このグラフは「ある現象を起こすには、約48 kcal/molのエネルギーが必要で、それを超えると変化が完了する」ことを示しています。
最後に
この手法はin silico創薬の論文の中で、よく見られる手法の一つです。
通常のリガンド-タンパク質のMDと比べると、少し難しいですが、その分行えばIFが高い雑誌に載せれそうな感じがしています。ぜひチャレンジしてみてください。
参考文献
Umbrella Sampling in Gromacs | MD Simulations with CHAPERONg
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

