

本記事は、タンパク質の構造予測、タンパク質-小分子の複合体構造を高精度に予測するAIツール Boltz について解説します。今回は、環境構築から使い方について、丁寧にご紹介します。
【この記事のまとめ】
創薬研究者やバイオインフォマティクスに携わる方へ。MITが開発したオープンソースのAIモデル「Boltz」を活用し、AlphaFold3と同等の高精度でタンパク質構造やタンパク質-リガンド複合体を予測・解析する方法を解説します。
- AlphaFold3並の精度をオープンソースで実現:拡散モデル(Diffusion Model)をベースとした最新アルゴリズムにより、バイオ分子複合体の三次元構造を高精度に予測。商用利用可能なMITライセンスで提供されています。
- 構造予測から親和性予測まで幅広く対応:アミノ酸配列のみの構造予測(Boltz-1)に加え、物理ベース計算(FEP)の1000倍以上の高速性で結合親和性を予測できるBoltz-2の可能性についても触れています。
- WSL2/GPU環境での実践的セットアップ:WSL2上での環境構築手順から、YAMLファイルを用いた具体的なタンパク質-リガンド複合体予測の実行コマンド、PyMOLでの可視化結果までを網羅しています。
この記事を読むことで、高額な商用ツールや制限のあるWebサービスに頼らず、自身のローカル/サーバー環境で自由度の高い「高精度バイオ分子シミュレーション」を完結させる手法が手に入ります。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700,
64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


Boltz とは?
Boltzは、MITの研究者らによって開発されたオープンソースのAIモデルで、バイオ分子複合体の三次元構造を高精度に予測します。特に、Google DeepMindのAlphaFold3と同等、あるいはそれ以上の精度を達成しながら、商用利用も可能なMITライセンスで提供されている点が大きな特徴です。
Boltzには、構造予測に特化した Boltz-1 と、それに加えて結合親和性予測も可能にした Boltz-2 が存在します。Boltz-2は、物理ベースの計算手法であるFEP (Free Energy Perturbation) と同等の精度を、なんと1000倍以上の高速性で実現するとされています。これにより、創薬における大規模な仮想スクリーニングが現実的な時間で行えるようになり、創薬研究を大きく加速させることが期待されています。
Boltz のアルゴリズム
Boltzのアルゴリズムは、基本的にAlphaFold3と同様に拡散モデル(Diffusion Model) をベースとしています。これは、ランダムなノイズから段階的に構造を「デノイズ(denoising)」していくことで、最終的な高精度な構造を生成するアプローチです。
入力データとして、アミノ酸配列(タンパク質)、SMILES文字列(小分子リガンド)、塩基配列(核酸)などを使用し、これらを統合的に学習します。さらに、Boltz-2では結合親和性を予測するための「アフィニティモジュール」が追加されており、構造予測と親和性予測を同時に行えるようになっています。
また、Boltzには「物理的誘導(Boltz-steering)」という技術が組み込まれており、これにより予測された構造に立体的な衝突などの非物理的なエラーがほとんど発生しないようになっています。
本記事では環境構築とタンパク質の構造予測、タンパク質ーリガンドの複合体予測を行います。
Boltz の利用法(コマンドラインでの利用)
Boltzは、GPU環境での利用が推奨されており、コマンドラインからの利用が一般的です。Webインターフェースを提供しているサービスもありますが、ここでは自身の環境で実行する方法を紹介します。
環境構築
環境構築をしていきます。今回はWindowsのWSLを利用します。GPUを利用するため、WSL2およびCUDAのセットアップが必要です。WSL2の利用方法については、過去の記事を参考にしてください。
以下のコマンドをWSL上で実行して、Boltzの環境を構築します。
# Boltzは、最新のPython環境にインストールすることを推奨
micromamba create -n boltz_env python=3.10 -y
micromamba activate boltz_env
# PyPIからBoltzをインストール
# GPU利用の場合は[cuda]を付ける。CPUのみの場合は削除
pip install boltz[cuda] -U
# またはGitHubから直接インストール
# git clone https://github.com/jwohlwend/boltz.git
# cd boltz
# pip install -e .[cuda]
コードの詳細説明
micromamba create -n boltz_env python=3.10 -y
micromamba を使用して、boltz_env という名前の新しい仮想環境を作成し、Python 3.10をインストールします。これにより、他のPythonプロジェクトとの依存関係の衝突を防ぐことができます。
micromamba activate boltz_env
作成した boltz_env 環境を有効化します。
pip install boltz[cuda] -U
pip を使用して、Python Package Index (PyPI) からBoltz本体をインストールします。
boltz[cuda]: GPUを利用するためのCUDA関連パッケージも同時にインストールします。U: パッケージを最新バージョンにアップグレードします。
これでBoltzを実行するための環境が整いました。
Boltzによるタンパク質構造予測
Boltzの実行は、タンパク質配列、小分子のSMILES文字列、またはPDBファイルなどを入力として行います。ここではPDB:9OB2のCDK2/CyclinEに化合物11が結合した結晶を例に行います。
YAMLファイルの作成
まずはBoltzを実行するための設定ファイルであるYAMLファイルを作成していきます。
まずは予測するタンパク質の配列を入手しましょう。
配列はPDBのfastaファイルに書いてあります。
ダウンロードしたFASTA Sequenceを開くと、以下の二つの配列があります。
>9OB2_1|Chain A|Cyclin-dependent kinase 2|Homo sapiens (9606)
SHMMENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL
>9OB2_2|Chain B|G1/S-specific cyclin-E1|Homo sapiens (9606)
GSIIAPSRGSPLPVLSWANREEVWKIMLNKEKTYLRDQHFLEQHPLLQPKMRAILLDWLMEVCEVYKLHRETFYLAQDFFDRYMATQENVVKTLLQLIGISSLFIAAKLEEIYPPKLHQFAYVTDGACSGDEILTMELMIMKALKWRLSPLTIVSWLNVYMQVAYLNDLHEVLLPQYPQQIFIQIAELLDLCVLDVDCLEFPYGILAASALYHFSSSELMQKVSGYQWCDIENCVKWMVPFAMVIRETGSSKLKHFRGVADEDAHNIQTHRDSLDLLDKARAKKA
これを元に以下のような感じでYAMLファイルを作成します。
詳しくはこちらをご覧ください。
cdk2_boltz_input.yaml という形で実装してください。
version: 1
sequences:
- protein:
id: A
sequence: SHMMENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL
- protein:
id: B
sequence: GSIIAPSRGSPLPVLSWANREEVWKIMLNKEKTYLRDQHFLEQHPLLQPKMRAILLDWLMEVCEVYKLHRETFYLAQDFFDRYMATQENVVKTLLQLIGISSLFIAAKLEEIYPPKLHQFAYVTDGACSGDEILTMELMIMKALKWRLSPLTIVSWLNVYMQVAYLNDLHEVLLPQYPQQIFIQIAELLDLCVLDVDCLEFPYGILAASALYHFSSSELMQKVSGYQWCDIENCVKWMVPFAMVIRETGSSKLKHFRGVADEDAHNIQTHRDSLDLLDKARAKKA
タンパク質構造予測の実行と結果
上記を用いて、例えば以下のコードを実行してください。
10分程度で解析が終わります。
boltz predict cdk2_boltz_input.yaml \
--use_msa_server \
--use_potentials \
--diffusion_samples 2 \
--recycling_steps 3 \
--output_format pdb \
--out_dir ./cdk2_boltz_out \
--override \
--no_kernels
各オプションの意味
-
boltz predict cdk2_boltz_input.yaml予測ジョブを実行。
cdk2_boltz_input.yamlに配列やテンプレ設定が書いてある前提。 -
-use_msa_serverColabFold のMSAサーバーで多重アラインメントを取得(外部アクセスあり)。ローカルMSAを使うなら外す。
-
-use_potentials幾何・物理的な拘束(ポテンシャル)を併用して、構造破綻を起きにくくするスイッチ。
-
-diffusion_samples 2拡散サンプル(独立生成)を2本だけ作る軽量設定。多様性は控えめ・速度は速め。
-
-recycling_steps 3リサイクル回数(精度↑だが計算↑)。まずは3で安定起動。
-
-output_format pdb出力をPDB形式で保存(mmCIFが良ければ
cif)。 -
-out_dir ./cdk2_boltz_out出力先ディレクトリ。
ranked_*.pdbやスコアJSON、ログが入る。 -
-override既存の出力があっても上書き。
-
-no_kernelsTritonの高速カーネルを使わずPyTorch実装にフォールバック(Ada世代での初期化エラー回避用)。速度はやや落ちるが安定。
実行後はpredictionsフォルダ に input_model_0.pdb などが生成されていれば成功しています。
pymolで可視化すると以下の感じです。
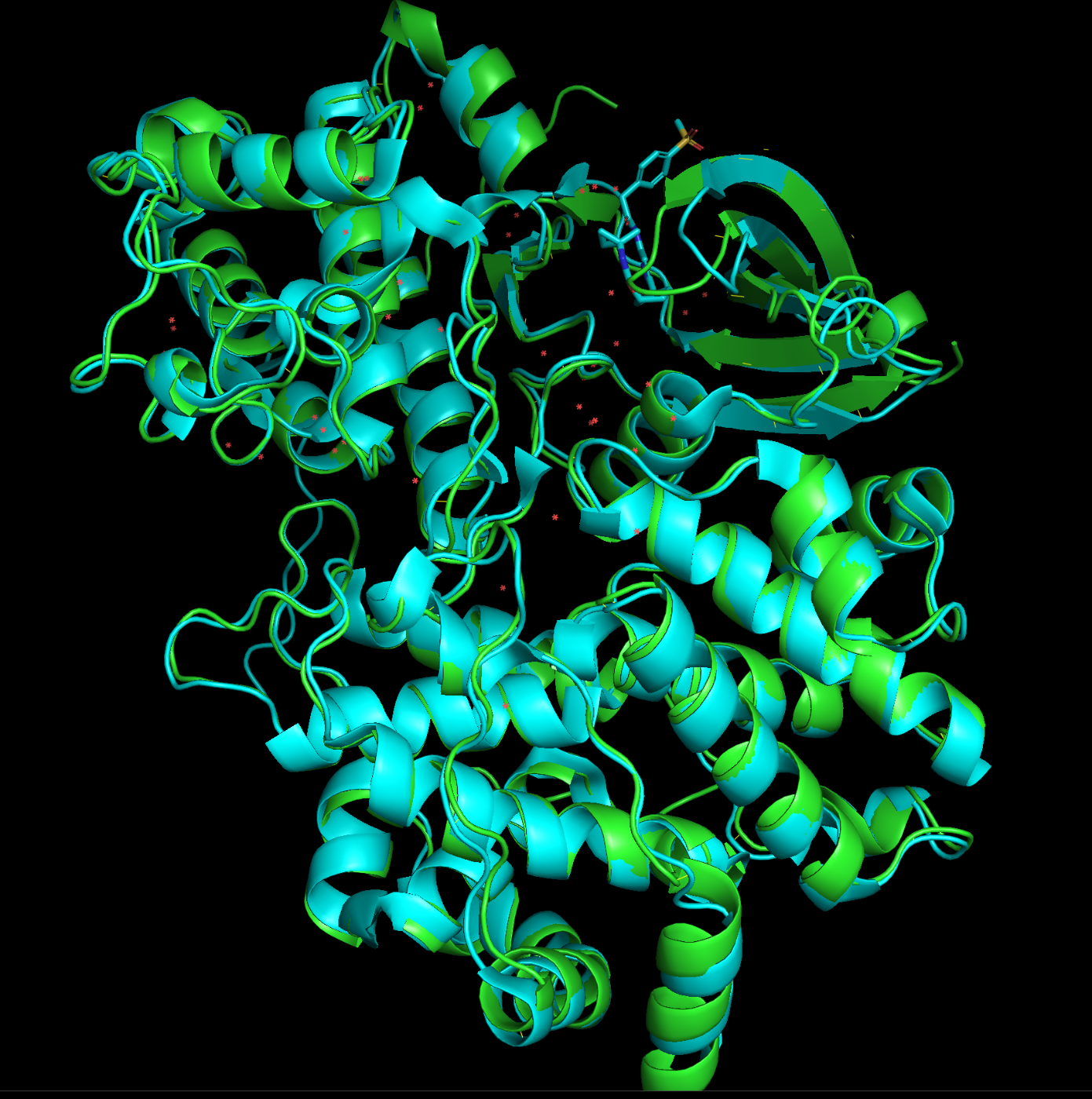
もともとのPDBと重ね合わせてみるとほぼ一致していることがわかります!
黄緑:Boltzで推測した構造
水色:もともとのPDB:9OB2
タンパク質-リガンド複合体の構造予測と結果
Boltzのgithubにyamlの例があるので、それを基に作成します。
yamlファイルを以下のように変更し、ligand_cdk2.yaml として保存してください。
version: 1 # Optional, defaults to 1
sequences:
- protein:
id: [A]
sequence: SHMMENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL
- ligand:
id: [B]
smiles: 'CC1(CC1)NC(=O)O[C@@H]2CC[C@@H](C2)c3[nH]nc(NC(=O)Cc4ccc(cc4)S(=O)(=O)C)c3'
inputのyamlファイルの箇所のみを変更し、以下を実行します。
boltz predict ligand_cdk2.yaml \
--use_msa_server \
--use_potentials \
--diffusion_samples 2 \
--recycling_steps 3 \
--output_format pdb \
--out_dir ./cdk2_boltz_out \
--override \
--no_kernels
実行後は同様のフォルダに結果cdk2_boltz_input_model_0.pdb が保存されています。
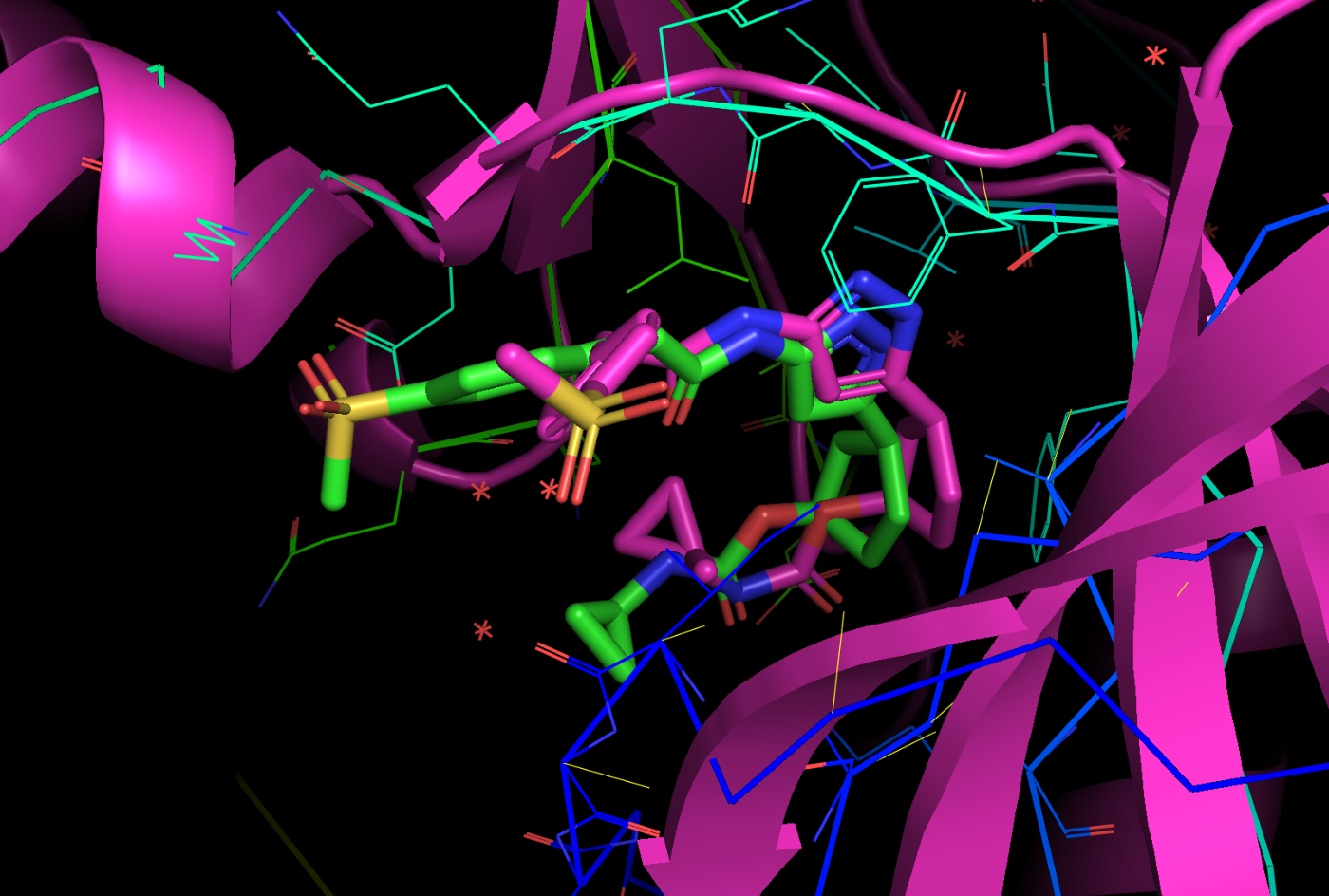
pymolで開くと以下のようになっています。確かにリガンドが結合していることがわかります。
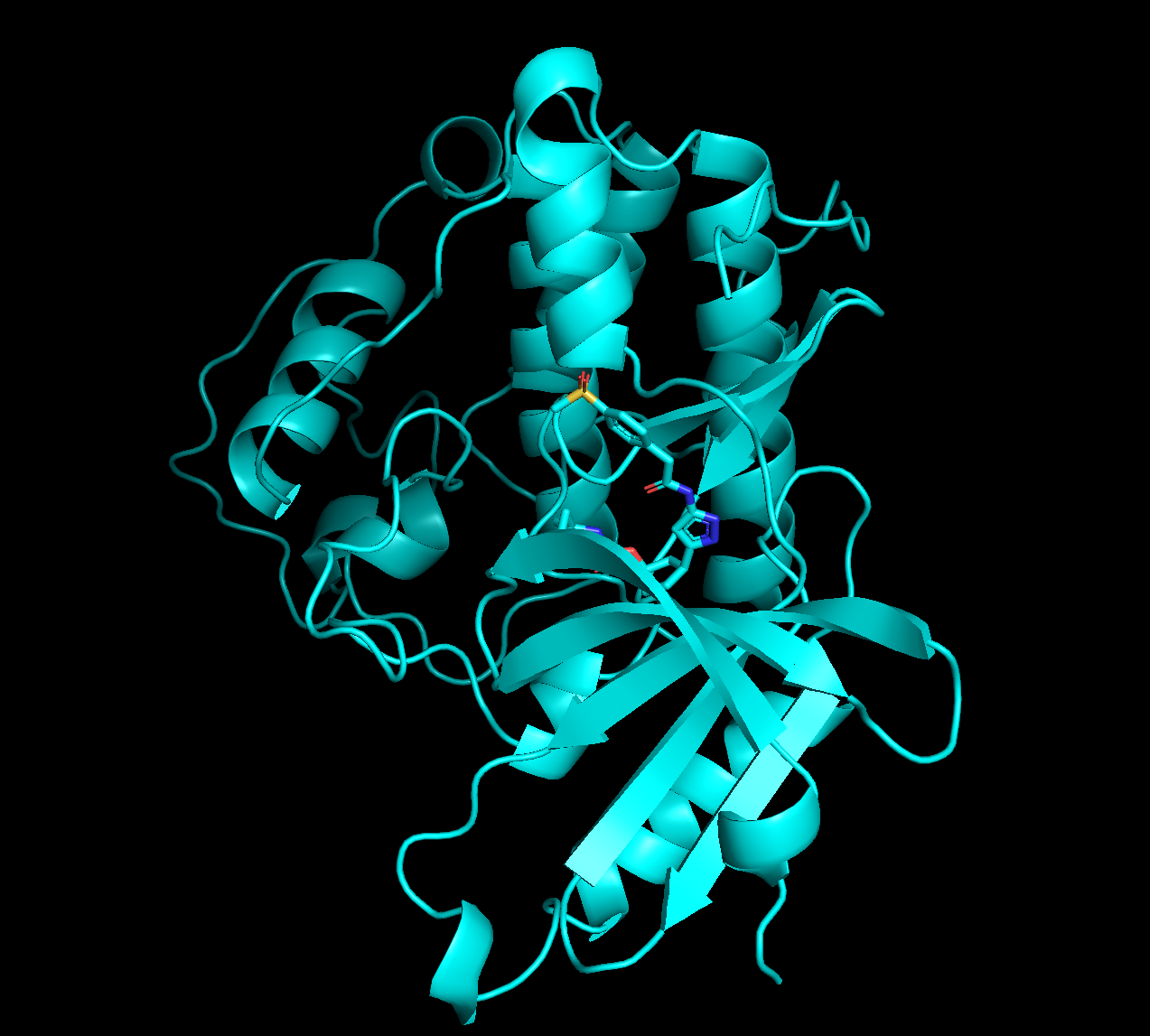
もともとのPDBと重ね合わせてみた結果が以下になります。
黄緑:Boltzで推測した構造
マゼンタ:もともとのPDB:9OB2
多少ずれていますが、同じポケットに結合しているのが予測できているのはすごいですね!
最後に
Boltzは、AlphaFold3と同等の高い精度を持ちながら、オープンソースで利用できる強力なツールです。今回は紹介しませんでしたが、Boltz2では結合力の予測や環状ペプチドの予測もできるようです。githubにyamlの書き方例があるので、ぜひ試してみてください! 環境構築は比較的シンプルで、GPUがあればすぐに始めることができます。ぜひ、ご自身の研究に取り入れてみてください。
Boltzのインストール方法や使い方について、さらに詳しい情報を知りたい場合は、Boltzの使い方を解説するチュートリアルビデオも参考にしてください。
参考文献
Boltz MIT License
GitHub – jwohlwend/boltz: Official repository for the Boltz biomolecular interaction models
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!
