

この記事は、Pythonで量子化学計算ができるPyscfを用いて、分子のIRスペクトルや熱力学的特性を取得する方法を解説します。DFT計算による構造最適化、振動数計算、IRスペクトルの可視化、熱力学的特性の取得までを扱います。これにより、Pyscfの物性解析を理解し、分子の最安定構造を得る方法を学ぶことができます。また、理論的なIRスペクトルと実験結果の比較による分子同定することが出来るようになります。
【この記事のまとめ】
計算化学の研究者や材料科学分野の学生に向けて、Pythonライブラリ「PySCF」を用いて赤外分光(IR)スペクトルを予測し、エンタルピーやギブス自由エネルギーなどの熱力学諸量を算出する具体的な手法を解説します。
- IRスペクトルの可視化:振動数解析から得られる強度データをもとに、実験値と比較可能な赤外分光チャートをPythonで描画するプロセスを習得できます。
- 統計力学に基づく熱力学計算:零点振動エネルギー(ZPE)の補正、エントロピー、ギブス自由エネルギーなど、反応の自発性や平衡定数の議論に不可欠な数値を導き出せます。
- 解析ワークフローの自動化:出力ファイルを手計算する手間を省き、計算実行からプロット作成までを一気通貫で自動化するスクリプトの実装方法を学べます。
この記事を読むことで、単なる構造最適化に留まらない「分子の熱力学的性質」の深い解析をPython上で完結させ、研究の精度と効率を大幅に向上させることができます。
Google colab, pyscf 2.5.0, py3Dmol


Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!


量子化学計算用のソフトウェア: PySCFとは
PySCF(Python Simulations of Chemistry Framework)は、Pythonで書かれた無料・商用利用が可能なオープンソースの量子化学計算Pythonライブラリです。PySCFは特に電子構造計算に対応しており、密度汎関数理論(DFT)、ハートリー・フォック法、ポストハートリー・フォック法など多様な計算手法をサポートしています。利用者はPythonを用いて容易に計算セットアップを行うことができ、拡張性とカスタマイズの自由度が高いため、研究開発に適しています。[1]。

参考文献[1]から引用
IRスペクトルと調和近似
1. 調和近似と振動数解析の役割
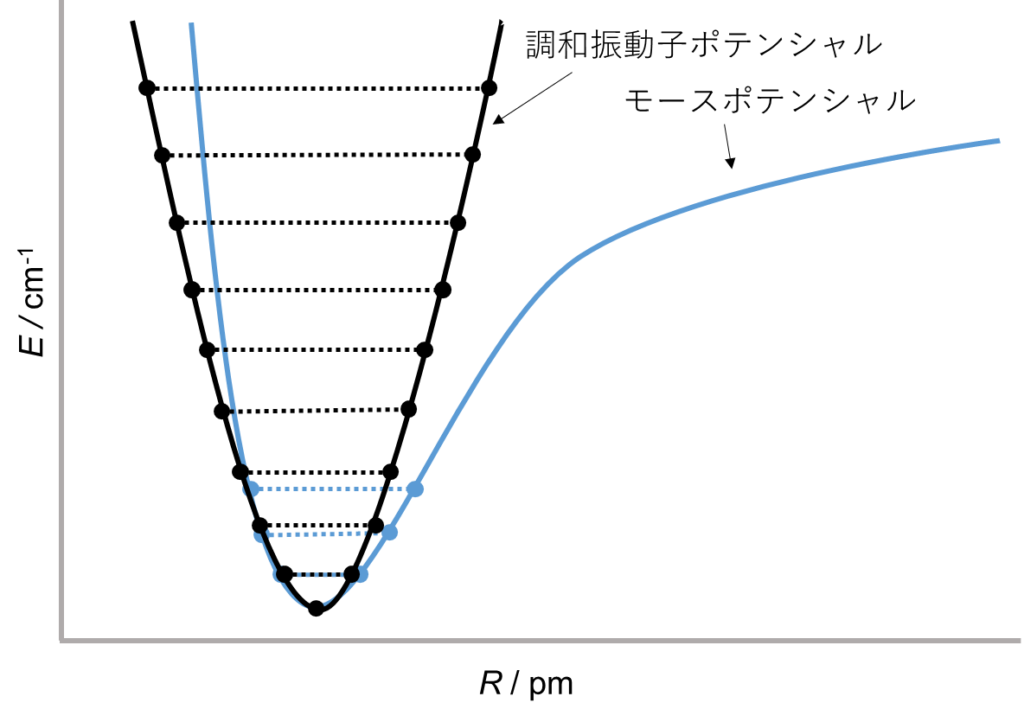
振動数解析は、構造最適化計算における構造が極小であるかどうかを確認するために行われます。具体的には、振動数解析では実際のポテンシャルを調和振動子による二次関数的なポテンシャルで近似しています。
平衡構造は、その放物線で表されるポテンシャル局面の頂点にある構造で、ばねで言えば力のかかっていない初期状態に対応します。これにより、構造が安定しているかどうかを確認することができます。
参考文献[2]
2. 計算手法と基底関数の影響
同じ分子でも、計算手法や基底関数を変えると平衡構造は多少変化します。また、X線構造解析など実験で得られる構造も、計算上で得られる平衡構造とは異なることがあります。そのため、異なる手法で計算した最適化構造や実験で得られた構造について、そのまま振動数解析を行うことには注意が必要です。
振動数解析を行う場合には、まず対象とする分子を構造最適化し、その最適化構造について、同じ手法と基底関数を用いて解析することが必要になります。
3. 調和近似の限界とスケール因子
調和近似によるポテンシャルの計算は簡便ですが、結合した原子間の相互作用をよりよく表すモースポテンシャルからは少しずれています。そのため、振動数解析から得られる振動数の計算値は、実際よりもずれを含んでいることを認識して利用する必要があります。
このずれを補正するために用いられるのがスケール因子です。
4. スケール因子とは
スケール因子は、振動数解析の計算値を実験値に合わせるための補正係数です。振動数の計算値が実際の測定値よりもずれている場合、このスケール因子を適用することで、より現実に近い値に補正することができます。スケール因子は、特定の計算手法や基底関数に基づいて経験的に決定されることが多く、文献[2]やこちらのサイトで提供されています。
| 6-31G* | 6-31G** | 6-31+G** | cc-pVDZ | cc-pVTZ | |
|---|---|---|---|---|---|
| HF | 0.899 | 0.903 | 0.904 | 0.908 | 0.91 |
| B3LYP | 0.96 | 0.961 | 0.964 | 0.97 | 0.967 |
| M06-2X | 0.947 | 0.95 | 0.952 | 0.955 | |
| PBEPBE | 0.986 | 0.986 | 0.989 | 0.994 | 0.993 |
| TPSSh | 0.959 | 0.959 | 0.963 | 0.972 | 0.968 |
| wB97X-D | 0.949 | 0.952 | 0.953 | 0.956 | |
| MP2 | 0.943 | 0.937 | 0.941 | 0.953 | 0.95 |
| CID | 0.924 | 0.924 | 0.924 | 0.924 | 0.927 |
以上の内容を基に、振動数解析の理解を深め、実際の計算に応用することが重要です。これにより、理論的なIRスペクトルと実験結果の比較が可能になり、分子同定や構造解析に役立てることができます。
PySCFを用いたIRスペクトルの計算手法
まず、対象となる分子の構造最適化計算を行っていきます。今回は1,2-ジフルオロエタンを使って計算していきます。具体的な構造最適化計算の方法はこちらの記事を参照してください。
今回はGoogle Colabを使って実行します。
Google Colabは、ブラウザベースのPythonプログラミング環境であり、PySCFを使用するのに適したプラットフォームの一つです。Google Colabを使用することで、ローカル環境のセットアップなしにPySCFの計算をすぐに開始することができます。
Google colabのリンク:https://colab.research.google.com/?hl=ja
1. 環境設定とインストール
以下のコードをGoogle Colabに入力して、必要なライブラリをインストールします。
!pip install pyscf
!pip install numpy
!pip install geometric
!pip install git+https://github.com/pyscf/properties
!pip install py3Dmol各ライブラリの説明
- PySCF: 量子化学計算のライブラリです。
- NumPy: 数値計算のライブラリです。
- geometric: 構造最適化を行うためのライブラリです。
- pyscf/properties: PySCFの拡張モジュールです。IRスペクトルの可視化に必要です。
- py3Dmol: 3D分子構造の可視化ライブラリです。
続いて、Google Driveをマウントし、計算結果を保存するディレクトリを設定します。
from google.colab import drive
drive.mount('/content/drive')
import os
# 計算結果を保存するパスを設定
folder_path = "/content/drive/MyDrive/pyscf_IR_thermo"
base_filename = "anticlinal_conformation"
# フルパスの設定
path = os.path.join(folder_path, base_filename)
# フォルダが存在しない場合は作成
if not os.path.exists(folder_path): os.makedirs(folder_path) print(f"Created folder: {folder_path}")
# 例としてチェックポイントファイル名の設定
checkpoint_file = path + ".chk"
print(f"Checkpoint file will be saved as: {checkpoint_file}")
2. 構造最適化
まず、対象分子の構造最適化を行います。今回は1,2-ジフルオロエタンを例に計算を進めます[3-7]。
from pyscf import gto
mol = gto.M( atom=''' F -2.9513875514 -1.0928382404 0.2476531628 F -5.6179262375 1.2415516978 -0.2476528977 C -3.5272115798 0.1385618493 -0.0055750514 C -5.0421022105 0.0101516046 0.0055753029 H -3.1869047581 0.5023934144 -0.9980363055 H -3.2040823737 0.8590234395 0.7752476523 H -5.3652314210 -0.7103099785 -0.7752474058 H -5.3824090296 -0.3536799681 0.9980365550 ''', basis='6-31G(d)', charge=0, spin=0, symmetry=True, verbose=4
)
from pyscf import dft
mf = dft.RKS(mol)
mf.chkfile = path + '.chk'
mf.xc = 'B3LYP'
mf.max_cycle = 150 # SCF計算の最大反復回数
energies = [] # 各ステップのエネルギーを保存するリスト
geometries = [] # 各ステップの分子構造を保存するリスト
# コールバック関数の定義
def cb(envs): mf = envs['g_scanner'].base if not hasattr(mf, 'e_tot'): print("エネルギーが更新されていません。") return energies.append(mf.e_tot) geometries.append(mol.atom_coords(unit="ANG")) print(f"ステップ {len(energies)}: エネルギー = {mf.e_tot} Hartree")
conv_params = { 'convergence_energy': 1e-6, # エネルギーの収束閾値 (Eh) 'convergence_grms': 3e-4, # 勾配のRMS収束閾値 (Eh/Bohr) 'convergence_gmax': 4.5e-4, # 最大勾配の収束閾値 (Eh/Bohr) 'convergence_drms': 1.2e-3, # 変位のRMS収束閾値 (Angstrom) 'convergence_dmax': 1.8e-3 # 最大変位の収束閾値 (Angstrom)
}
from pyscf.geomopt import geometric_solver
import matplotlib.pyplot as plt
# 構造最適化
geometric_solver.optimize(mf, callback=cb, **conv_params)
# エネルギー収束のプロット
plt.figure(figsize=(10, 5))
plt.plot(energies, marker='o')
plt.title('Energy Convergence During Geometry Optimization')
plt.xlabel('Optimization Step')
plt.ylabel('Total Energy (Hartree)')
plt.grid(True)
plt.show()
# ジオメトリーの保存
for i, coords in enumerate(geometries): filename = f"{path}_geometry_step_{i+1}.xyz" with open(filename, 'w') as f: f.write(f"{mol.natm}\\\\n") f.write(f"Step {i+1} Energy: {energies[i]}\\\\n") for j in range(mol.natm): symbol = mol.atom_pure_symbol(j) x, y, z = coords[j] f.write(f"{symbol} {x:.6f} {y:.6f} {z:.6f}\\\\n")
print("All geometries have been saved.")
3. 分子構造の可視化
最適化された分子構造を3Dで可視化します[8]。
import ipywidgets
from ipywidgets import interact
import py3Dmol
# モデルのXYZ座標をファイルから読み込む
def read_xyz(file_path): with open(file_path, 'r') as file: return file.read()
# py3Dmolでのビューアを設定する関数
def show_structure(xyz_content, style): view = py3Dmol.view(width=800, height=400) view.addModel(xyz_content, 'xyz') if style == 'ball and stick': view.setStyle({'stick':{}, 'sphere':{'radius': 0.5}}) elif style == 'stick': view.setStyle({'stick': {}}) elif style == 'line': view.setStyle({'line': {'linewidth': 5}}) elif style == 'sphere': view.setStyle({'sphere': {}}) view.setBackgroundColor('white') view.zoomTo() return view.show()
# ファイルパスを設定
filename_template = f"{path}_geometry_step_"
# 最適化ステップの数に応じたインタラクション
def style_selector(idx, s): filename = f"{filename_template}{idx}.xyz" xyz_content = read_xyz(filename) show_structure(xyz_content, s)
# インタラクションウィジェットの設定
max_steps = i + 1 # 最適化ステップの総数
interact(style_selector, idx=ipywidgets.IntSlider(min=1, max=max_steps, step=1, value=max_steps, description='Step'), s=ipywidgets.Dropdown( options=['ball and stick', 'stick','line', 'sphere'], value='ball and stick', description='Style:'))
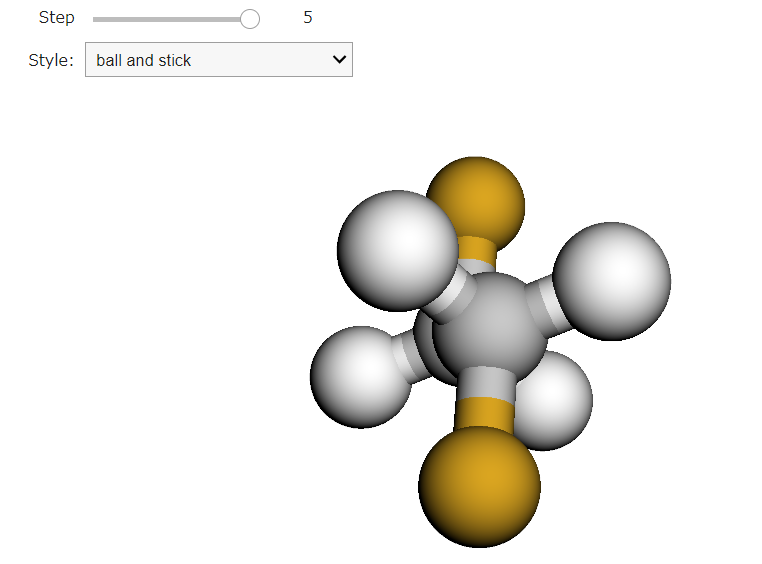
この結果、以下のようにアンチ型の1,2-ジフルオロエタンを得ることが出来ました。
4. 振動数解析とIRスペクトルの取得
振動数解析は、分子の構造がエネルギー極小点であるかを確認し、IRスペクトルを取得するために行います。以下のコードを使用して振動数解析とIRスペクトルの計算を行います。
import matplotlib.pyplot as plt
import sys
from pyscf import gto, dft, lib
from pyscf.hessian import rks # 振動数計算のためのモジュール
from pyscf.prop import infrared
from pyscf.hessian import thermo
# チェックポイントファイルから分子情報をロード
# 'path'はチェックポイントファイルのパスです。チェックポイントファイルには、前回の計算結果が保存されています。
mol = lib.chkfile.load_mol(path + '.chk')
# DFT計算の設定
# チェックポイントファイルを指定して、前回の計算結果を引き継ぎます。
mf = dft.RKS(mol)
mf.chkfile = path + '.chk' # チェックポイントファイルの指定
mf.xc = 'b3lyp' # 使用する交換・相関汎関数の指定
# DFT計算の実行
# 分子の電子構造を計算します。
mf.kernel()
# IRスペクトル解析の実行
# infraredモジュールを使用して赤外スペクトルを計算します。
mf_ir = infrared.rks.Infrared(mf).run()
# IRスペクトルの概要を表示
# 計算結果のサマリーを表示します。
mf_ir.summary()
# 熱力学的計算の実行
# thermoモジュールを使用して、計算した振動周波数を基に熱力学的性質を評価します。
thermo.dump_thermo(mol, thermo.thermo(mf, mf_ir.vib_dict["freq_au"], 298.15, 101325))
# IRスペクトルのプロット
# 振動数のスケーリングファクターとして0.960を適用
# 詳細はCCCBDBの推奨を参照(<https://cccbdb.nist.gov/vibscalejust.asp)>
fig, ax, ax2 = mf_ir.plot_ir(w=100, scale=0.960)
# プロットのタイトルを設定
# プロットの見出しを設定し、詳細を含めます。
ax.set_title(r"Infrared Spectra of Methylformate Molecule ($\\\\\\\\mathrm{CH2FCH2F}$)" "\\\\\\\\n" r"B3LYP/6-31G(d) with scaling factor 0.960 and Lorentz broadening with $\\\\\\\\mathrm{FWHW = 100 cm^{-1}}")
# レイアウトの調整とプロットの表示
# プロットのレイアウトを調整して表示します。
fig.tight_layout()
fig.show()
詳細説明
- モジュールのインポート:
matplotlib.pyplot:プロットを作成するためのモジュール。sys:システム関連の操作を行うためのモジュール。pyscf.gto、pyscf.dft、pyscf.lib:PySCFの分子構造と密度汎関数理論(DFT)計算を行うためのモジュール。pyscf.hessian.rks:DFTで振動数計算を行うためのモジュール。pyscf.prop.infrared:赤外スペクトル解析を行うためのモジュール。pyscf.hessian.thermo:熱力学的計算を行うためのモジュール。
- 分子のロード:
- チェックポイントファイルから分子情報をロードします。チェックポイントファイルには上で構造最適化した計算結果が保存されています。
- DFT計算の設定:
- チェックポイントファイルを指定し、構造最適化の計算結果を引き継いでDFT計算を行います。使用する交換・相関汎関数としてB3LYPを指定します。
- DFT計算の実行:
- 分子の電子構造を計算します。
- IRスペクトル解析の実行:
infraredモジュールを使用して赤外スペクトルを計算し、結果の概要を表示します。
- 熱力学的計算の実行:
- 計算した振動周波数を基に、298.15Kと標準状態の圧力(101325Pa)での熱力学的性質を評価します。
- IRスペクトルのプロット:
- 振動数のスケーリングファクターとして0.960を適用し、半値全幅を100cm⁻¹としてIRスペクトルをプロットします。
- プロットにタイトルを設定し、結果を表示します。
このようにして得られたIRスペクトルは以下の画像となり、実験データとの比較により、分子の同定や構造解析に役立ちます。
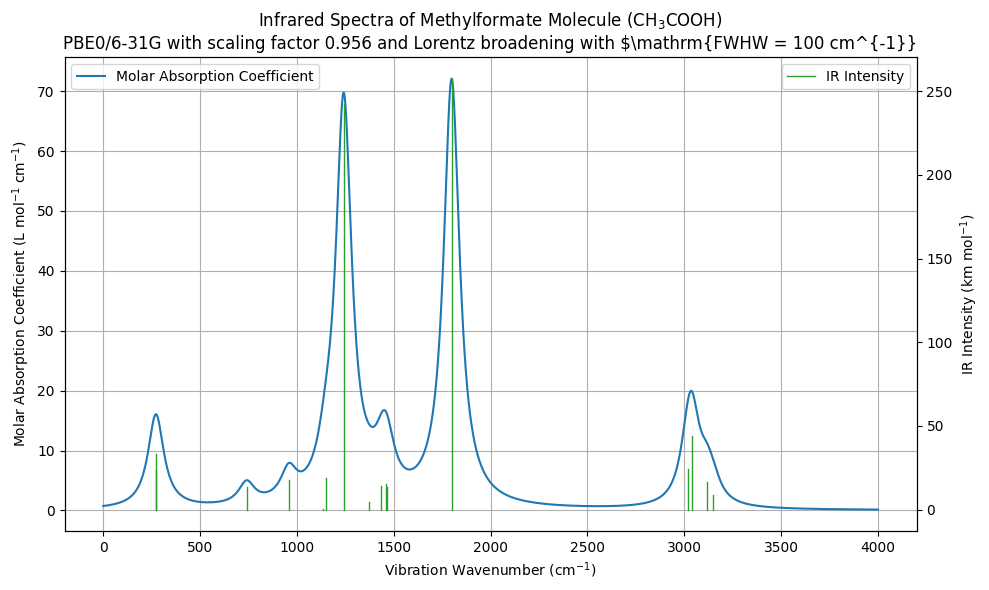
このセクションでは、1,2-ジフルオロエタンの振動数解析とIRスペクトルの取得方法を解説しました。PySCFを用いて構造最適化を行い、その後に振動数解析を行うことで、分子の安定性を確認し、IRスペクトルを生成することができます。このプロセスにより、理論計算と実験データを比較し、分子の特性を深く理解することが可能です。
熱力学的特性と異性体の全エネルギー差
1. 熱力学的特性とは
熱力学的特性とは、物質のエネルギー状態や反応性を示す一連の物理的性質を指します。これには、エンタルピー(H)、エントロピー(S)、全エネルギー(E)などが含まれます。これらの特性は、化学反応や物理変化が起こる際のエネルギーの流れや平衡状態を理解するために重要です。
2. 全エネルギー(E)
全エネルギー(E)は、系の内部エネルギーを示す指標です。全エネルギーには、システムのポテンシャルエネルギーや運動エネルギーが含まれます。化学反応や物理変化が起こる際には、全エネルギーの変化が重要な役割を果たします。
3. 異性体の全エネルギー差から分かること
異性体とは、同じ分子式を持ちながら異なる構造を持つ化合物のことです。異性体間の全エネルギー差を比較することで、どの異性体が最も安定しているかを判断することができます。
例えば、異性体Aと異性体Bの全エネルギーをそれぞれ $E_A$と $E_B$とします。このとき、全エネルギー差 $\Delta E$ は次のように定義されます:
$$ \Delta E = E_B – E_A $$
この値が負であれば、異性体Aが異性体Bよりも安定していることを示します。逆に、正の値であれば、異性体Bがより安定していることを示します。
熱力学的特性と全エネルギーの理解は、化学反応や物質の安定性を評価するために不可欠です。異性体の全エネルギー差を比較することで、どの異性体が最も安定しているかを判断できるため、化学研究や材料科学において重要な指標となります。量子化学計算を通じてこれらの特性を詳しく解析することで、実験結果との比較や新しい分子の設計に活用することができます。
異性体のエネルギー計算
今回は1,2-ジフルオロエタンの構造異性体であるゴーシュ型の分子を計算し、先ほど計算したアンチ型の1,2-ジフルオロエタンとエネルギーの比較を行います。まず、新しいフォルダを作成します。
import os
# 計算結果を保存するパスを設定
folder_path = "/content/drive/MyDrive/pyscf_IR_thermo"
base_filename2 = "gauche_conformation"
# フルパスの設定
path2 = os.path.join(folder_path, base_filename2)
# フォルダが存在しない場合は作成
if not os.path.exists(folder_path): os.makedirs(folder_path) print(f"Created folder: {folder_path}")
# 例としてチェックポイントファイル名の設定
checkpoint_file2 = path2 + ".chk"
print(f"Checkpoint file will be saved as: {checkpoint_file2}")
続いて、ゴーシュ型異性体のエネルギー計算を行います。
import matplotlib.pyplot as plt
import sys
from pyscf import gto, dft, lib
from pyscf.geomopt import geometric_solver
from pyscf.hessian import rks # 振動数計算のためのモジュール
from pyscf.hessian import thermo
# 分子の定義
mol = gto.M( atom=''' F -2.9646670035 1.3527250959 0.1781513936 F -4.6216795179 -0.7305791160 -0.6508177073 C -4.2423816872 1.5905363784 -0.2946175127 C -5.1302460519 0.3878960539 -0.0160002927 H -4.1984219564 1.7820726269 -1.3875645139 H -4.6566700057 2.4872569027 0.2125627625 H -5.1771329542 0.1985823306 1.0772122356 H -6.1561483328 0.5846824999 -0.3925288715 ''', basis='6-31G(d)', verbose=4
)
# DFT計算の設定
mf = dft.RKS(mol)
mf.chkfile = checkpoint_file2
mf.xc = 'B3LYP'
mf.max_cycle = 150 # SCF計算の最大反復回数
energies = [] # 各ステップのエネルギーを保存するリスト
geometries = [] # 各ステップの分子構造を保存するリスト
# コールバック関数の定義
def cb(envs): mf = envs['g_scanner'].base if not hasattr(mf, 'e_tot'): print("エネルギーが更新されていません。") return energies.append(mf.e_tot) geometries.append(mol.atom_coords(unit="ANG")) print(f"ステップ {len(energies)}: エネルギー = {mf.e_tot} Hartree")
conv_params = { 'convergence_energy': 1e-6, # エネルギーの収束閾値 (Eh) 'convergence_grms': 3e-4, # 勾配のRMS収束閾値 (Eh/Bohr) 'convergence_gmax': 4.5e-4, # 最大勾配の収束閾値 (Eh/Bohr) 'convergence_drms': 1.2e-3, # 変位のRMS収束閾値 (Angstrom) 'convergence_dmax': 1.8e-3 # 最大変位の収束閾値 (Angstrom)
}
# 構造最適化
geometric_solver.optimize(mf, callback=cb, **conv_params)
# エネルギー収束のプロット
plt.figure(figsize=(10, 5))
plt.plot(energies, marker='o')
plt.title('Energy Convergence During Geometry Optimization')
plt.xlabel('Optimization Step')
plt.ylabel('Total Energy (Hartree)')
plt.grid(True)
plt.show()
# ジオメトリーの保存
for i, coords in enumerate(geometries): filename = f"{path2}_geometry_step_{i+1}.xyz" with open(filename, 'w') as f: f.write(f"{mol.natm}\\\\n") f.write(f"Step {i+1} Energy: {energies[i]}\\\\n") for j in range(mol.natm): symbol = mol.atom_pure_symbol(j) x, y, z = coords[j] f.write(f"{symbol} {x:.6f} {y:.6f} {z:.6f}\\\\n")
print("All geometries have been saved.")
ゴーシュ型の分子構造が得られたことを確認し、可視化します。
import ipywidgets
from ipywidgets import interact
import py3Dmol
# モデルのXYZ座標をファイルから読み込む
def read_xyz(file_path): with open(file_path, 'r') as file: return file.read()
# py3Dmolでのビューアを設定する関数
def show_structure(xyz_content, style): view = py3Dmol.view(width=800, height=400) view.addModel(xyz_content, 'xyz') if style == 'ball and stick': view.setStyle({'stick':{}, 'sphere':{'radius': 0.5}}) elif style == 'stick': view.setStyle({'stick': {}}) elif style == 'line': view.setStyle({'line': {'linewidth': 5}}) elif style == 'sphere': view.setStyle({'sphere': {}}) view.setBackgroundColor('white') view.zoomTo() return view.show()
# ファイルパスを設定
filename_template = f"{path2}_geometry_step_"
# 最適化ステップの数に応じたインタラクション
def style_selector(idx, s): filename = f"{filename_template}{idx}.xyz" xyz_content = read_xyz(filename) show_structure(xyz_content, s)
# インタラクションウィジェットの設定
max_steps = i + 1 # 最適化ステップの総数
interact(style_selector, idx=ipywidgets.IntSlider(min=1, max=max_steps, step=1, value=max_steps, description='Step'), s=ipywidgets.Dropdown( options=['ball and stick', 'stick','line', 'sphere'], value='ball and stick', description='Style:'))
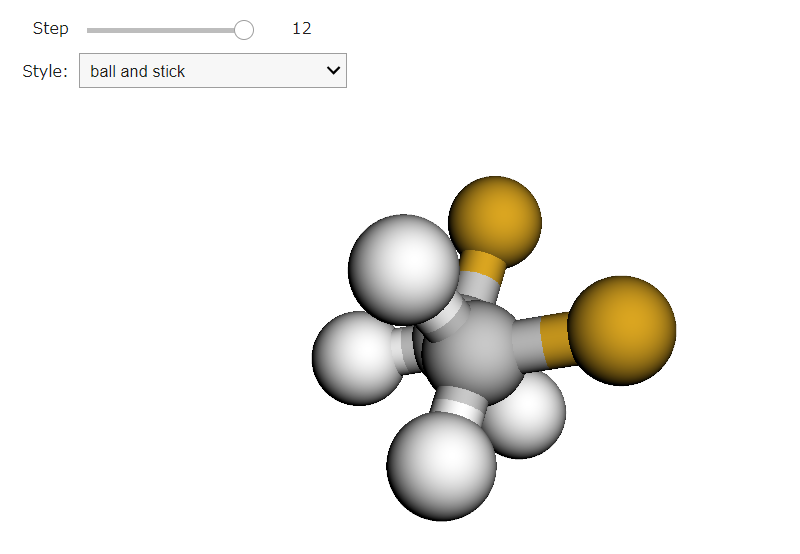
この結果、ゴーシュ型の分子が得られていることがわかります。
異性体のギブズエネルギー差の計算と比較
それでは1,2-ジフルオロエタンの異性体の熱力学的特性を計算し、異性体間のギブズエネルギー差を比較して行きます。
必要なライブラリのインポート
import pandas as pd
import os
from pyscf import gto, dft, lib
from pyscf.hessian import rks, thermoまず、計算に必要なライブラリをインポートします。
定数定義
# Hartree to eV conversion factor
HARTREE_TO_EV = 27.2114ハートリーを電子ボルト(eV)に変換するための定数を定義します。
熱化学特性を計算する関数
def calculate_thermo_properties(path): # Checkpointファイルから分子情報をロード mol = lib.chkfile.load_mol(path + '.chk') # DFT計算の設定 mf = dft.RKS(mol) mf.chkfile = path + '.chk' mf.xc = 'b3lyp' # DFT計算 mf.kernel() # 振動数計算の実行 hessian = rks.Hessian(mf) freqs = hessian.kernel() # Thermochemistry analysis at 298.15 K and 1 atmospheric pressure freq_info = thermo.harmonic_analysis(mf.mol, freqs) thermo_info = thermo.thermo(mf, freq_info['freq_au'], 298.15, 101325) # 熱化学特性を辞書形式で返す return { "File Name": os.path.basename(path), "Imaginary Freq (cm^-1)": freq_info['freq_error'], "Temperature (K)": thermo_info['temperature'][0], "Pressure (atm)": thermo_info['pressure'][0] / 101325, # Pa to atmに変換 "Frequencies scaled by": 1.0000, "Electronic Energy (EE) (Hartree)": thermo_info['E0'][0], "Zero-point Energy Correction (Hartree)": thermo_info['ZPE'][0], "Thermal Correction to Energy (Hartree)": thermo_info['E_vib'][0], "Thermal Correction to Enthalpy (Hartree)": thermo_info['H_vib'][0], "Thermal Correction to Free Energy (Hartree)": thermo_info['G_vib'][0], "EE + Zero-point Energy (Hartree)": thermo_info['E_0K'][0], "EE + Thermal Energy Correction (Hartree)": thermo_info['E_tot'][0], "EE + Thermal Enthalpy Correction (Hartree)": thermo_info['H_tot'][0], "EE + Thermal Free Energy Correction (Hartree)": thermo_info['G_tot'][0], "E (Thermal) (Hartree)": thermo_info['E_0K'][0], # Thermal energyとして仮定 "Heat Capacity (Cv) (J/mol·K)": thermo_info['Cv_tot'][0], "Entropy (S) (J/mol·K)": thermo_info['S_tot'][0] }calculate_thermo_properties関数は、指定されたパスに基づいて分子のチェックポイントファイルをロードし、DFT計算を実行して熱化学特性を計算します。計算結果は辞書形式で返されます。
計算結果の取得と表示
# 最初の構造の計算結果
thermo_properties_1 = calculate_thermo_properties(path)
# 2番目の構造の計算結果
thermo_properties_2 = calculate_thermo_properties(path2)
# データフレームに追加
df = pd.DataFrame([thermo_properties_1, thermo_properties_2])
# 最小エネルギーを持つ構造を見つける
min_energy = df["EE + Zero-point Energy (Hartree)"].min()
# 相対エネルギーを計算し、eV単位に変換
df["Relative Energy (eV)"] = (df["EE + Zero-point Energy (Hartree)"] - min_energy) * HARTREE_TO_EVまず、2つの分子構造の計算結果を取得し、それぞれをcalculate_thermo_properties関数で計算します。計算結果はpandasのDataFrameに追加され、最小エネルギーを持つ構造を見つけ、相対エネルギーを計算してeV単位に変換します。
行と列を入れ替えて表示
# 行と列を入れ替える
transposed_df_with_units = df.set_index("File Name").T
# 結果を表示
transposed_df_with_units最終的に、DataFrameの行と列を入れ替え、単位付きの計算結果を表示します。
このスクリプトは、異なる分子構造の熱化学特性を計算し、それらを比較するのに役立ちます。PySCFを使用してDFT計算を行い、計算結果をpandasのDataFrameに格納し、さらに見やすい形式で表示するために行と列を入れ替えています。最後の行を見ることで、計算した分子の中で最も安定な分子構造から他の異性体がどれだけ不安定か分かり、最安定構造と異性体の熱力学的特性を比較することができます。
今回は良く用いられるゼロ点エネルギー補正された全電子エネルギー(EE + Zero-point Energy (Hartree))eVに変換して比較しました。
ゼロ点振動エネルギーとは、量子力学的に許される最も低い振動エネルギーを表し、温度が絶対零度であっても振動モードが持つエネルギーです。
| File Name | anticlinal conformation | gauche conformation |
|---|---|---|
| Imaginary Freq | 0.000000 | 0.000000 |
| Temperature (K) | 298.150000 | 298.150000 |
| Pressure (atm) | 1.000000 | 1.000000 |
| Frequencies scaled by | 1.000000 | 1.000000 |
| Electronic Energy (EE) (Hartree) | -278.274171 | -278.274766 |
| Zero-point Energy Correction (Hartree) | 0.061572 | 0.061503 |
| Thermal Correction to Energy (Hartree) | 0.063175 | 0.062977 |
| Thermal Correction to Enthalpy (Hartree) | 0.063175 | 0.062977 |
| Thermal Correction to Free Energy (Hartree) | 0.060405 | 0.060536 |
| EE + Zero-point Energy (Hartree) | -278.212599 | -278.213264 |
| EE + Thermal Energy Correction (Hartree) | -278.208164 | -278.208957 |
| EE + Thermal Enthalpy Correction (Hartree) | -278.207220 | -278.208012 |
| EE + Thermal Free Energy Correction (Hartree) | -278.239070 | -278.240265 |
| E (Thermal) (Hartree) | -278.212599 | -278.213264 |
| Heat Capacity (Cv) (J/mol·K) | 0.000021 | 0.000021 |
| Entropy (S) (J/mol·K) | 0.000107 | 0.000108 |
| Relative Energy (eV) | 0.018080 | 0.000000 |
以上のコードにより、異性体の熱力学的特性とギブズエネルギー差を比較することができ、どの異性体が最も安定しているかを判断することができます。
Gaussianとの比較
今回、1,2-ジフルオロエタンの構造異性体のギブズエネルギー差をPySCFを用いて計算し、その結果をGaussianでの計算結果と比較しました。
PySCFでの計算結果
PySCFを用いて1,2-ジフルオロエタンの構造異性体のギブズエネルギー差を計算しました。最小エネルギーを持つ異性体を基準に、他の異性体の相対エネルギーを求めたところ、相対エネルギーは0.0181 eVとなりました。
Gaussian16での計算結果
次に、同じ分子についてGaussianを用いて計算を行いました。Gaussianは、量子化学計算の分野で広く使用されている商用ソフトウェアであり、信頼性の高い計算結果を提供します。Gaussianで得られた相対エネルギーは0.0163 eVでした。
PySCFとGaussianの比較
PySCFとGaussianで得られた相対エネルギーの差は0.0018 eVでした。この値は非常に小さいため、PySCFの計算結果が信頼性のあるものであることがわかります。
終わりに
いかがでしたでしょうか。PySCFはGoogle colab上でIRスペクトルの可視化が出来きます。さらに、有料版などを使うと自宅PCではできないような大きな分子も計算可能です。是非気軽に色んな分子を試してみてください!
参考文献
[1] PySCF 2.4 公式サイト, https://pyscf.org/, (参照2024-05-27)[2] 西長亨、本田康 「有機化学者のための量子化学計算入門 Gaussianの基本と有効利用のヒント」 P. 110-113[3] Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Koval, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Scheurer, H. F. Schurkus, J. E. T. Smith, C. Sun, S.-N. Sun, S. Upadhyay, L. K. Wagner, X. Wang, A. White, J. Daniel Whitfield, M. J. Williamson, S. Wouters, J. Yang, J. M. Yu, T. Zhu, T. C. Berkelbach, S. Sharma, A. Yu. Sokolov, and G. K.-L. Chan, Recent developments in the PySCF program package, J. Chem. Phys. 153, 024109 (2020)[4] Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. McClain, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: the Python-based simulations of chemistry framework, WIREs Comput. Mol. Sci. 8, e1340 (2018)[5]Ulf Ekström, Lucas Visscher, Radovan Bast, Andreas J. Thorvaldsen, and Kenneth Ruud, Arbitrary-order density functional response theory from automatic differentiation, J. Chem. Theory Comput. 6, 1971 (2010).[6] Susi Lehtola, Conrad Steigemann, Micael J.T. Oliveira, and Miguel A.L. Marques, Recent developments in libxc — A comprehensive library of functionals for density functional theory, SoftwareX 7, 1 (2018).[7] L.-P. Wang, C.C. Song, “Geometry optimization made simple with translation and rotation coordinates”, J. Chem, Phys. 144, 214108 (2016).[8] py3Dmol in Jupyter, https://birdlet.github.io/2019/10/02/py3dmol_example/ (参照2024-05-27)[9] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, J. W. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT (2016).
Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

