

この記事では、Pythonで量子化学計算ができるPySCFを用いて、分子の分子軌道を取得・可視化する方法を解説します。特に、DFT計算による構造最適化、振動数計算、分子軌道の可視化手順について詳述します。これにより、PySCFを使用して簡単に対象分子の分子軌道を得ることができ、分子軌道の解析を通じて化学反応性の予測が可能になります。
【この記事のまとめ】
化学・材料科学の研究者や計算化学を学ぶ方に向けて、Pythonの「PySCF」ライブラリを活用し、分子の反応性や物性の鍵となる「分子軌道(MO)」を計算・可視化する方法を解説します。
- HOMO/LUMOの特定とエネルギー解析:分子の電子供与性・受容性を決める最高被占軌道(HOMO)と最低空軌道(LUMO)のエネルギー値を抽出し、反応性を定量的に評価する手法を学べます。
- 電子密度分布の3D視覚化:PySCFで計算したデータからCubeファイルを生成し、
py3Dmol等を用いてブラウザ上でインタラクティブに分子軌道の形状を確認するワークフローを紹介します。 - プログラミングによる解析の自動化:GUIソフトの操作に依存せず、多数の分子に対して分子軌道の形状やエネルギーギャップを一括で解析・画像保存する効率的な環境を構築できます。
この記事を読むことで、目に見えないミクロな電子の振る舞いをPythonで「見える化」し、化学反応のメカニズム解明や材料設計に役立てるスキルが身につきます。
Google colab, pyscf 2.5.0, py3Dmol


Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!


量子化学計算用のソフトウェア: PySCFとは
PySCF(Python Simulations of Chemistry Framework)は、Pythonで書かれた無料・商用利用が可能なオープンソースの量子化学計算Pythonライブラリです。PySCFは特に電子構造計算に対応しており、密度汎関数理論(DFT)、ハートリー・フォック法、ポストハートリー・フォック法など多様な計算手法をサポートしています。利用者はPythonを用いて容易に計算セットアップを行うことができ、拡張性とカスタマイズの自由度が高いため、研究開発に適しています。[1]。

参考文献[1]から引用
1. 分子軌道とは?
1.1 分子軌道の定義
分子軌道(Molecular Orbital, MO)は、分子内の電子の存在確率を記述する数学的関数です。分子軌道は、個々の原子軌道が結合または反結合して形成されます。分子軌道理論は、分子全体の電子構造を理解するための重要な手法であり、化学結合の性質や反応性を説明する際に用いられます。
1.2 原子軌道と分子軌道の違い
原子軌道は、単一の原子に属する電子の存在確率を示す関数であり、s軌道、p軌道、d軌道などがあります。これに対して、分子軌道は複数の原子の原子軌道が重なり合って形成され、分子全体の電子の振る舞いを記述します。具体的には、原子軌道が線形結合(線形結合分子軌道法、LCAO)して分子軌道が形成されます。
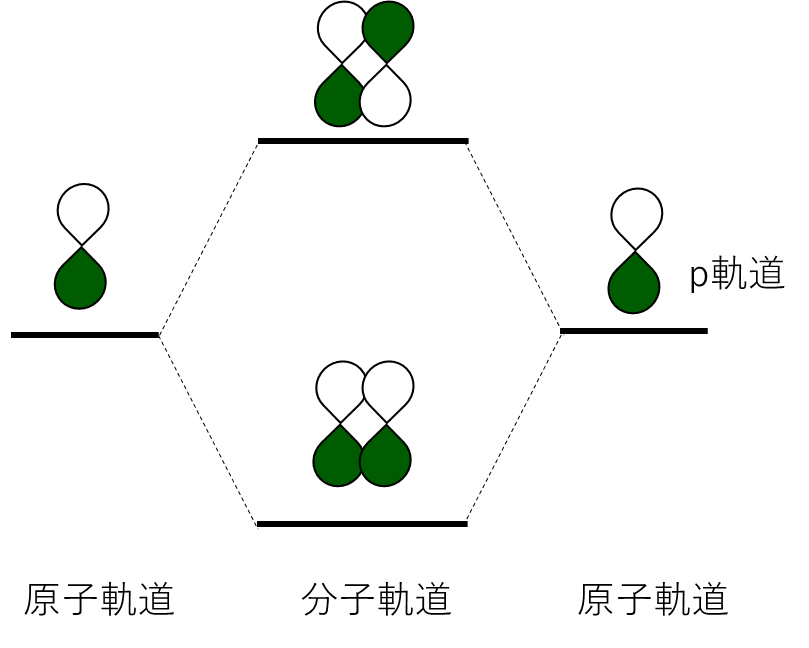
1.3 分子軌道の種類
分子軌道は、結合軌道(Bonding Orbital)と反結合軌道(Antibonding Orbital)、および非結合軌道(Non-bonding Orbital)に分類されます。
- 結合軌道: 結合軌道は、原子軌道が重なり合い、エネルギーが低下することで形成されます。結合軌道に電子が存在することで、原子同士の結合が強化されます。
- 反結合軌道: 反結合軌道は、電子によって占有された場合に2つの原子間の結合を弱め、分かれた原子の状態よりも分子のエネルギーを上昇させる分子軌道です。このような軌道は核間の結合領域に1つ以上の節を持ちます。
- 非結合軌道: 非結合軌道は、ある原子の原子軌道が他の原子と結合せずに独立して存在する場合に形成されます。この軌道の電子は、結合の強さに直接影響を与えません。
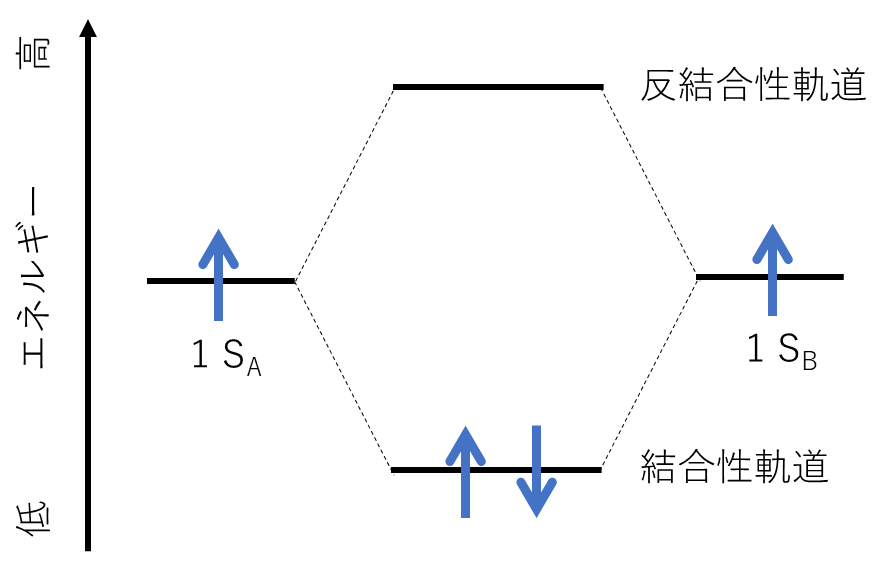
1.4 分子軌道理論の基本概念
分子軌道理論は、分子内の電子が特定の軌道に存在し、それぞれの軌道が異なるエネルギーを持つことを前提としています。この理論により、分子の電子配置や化学反応における電子の移動を理解することが可能になります。特に、分子軌道理論は以下の点で重要です。
- エネルギー準位: 各分子軌道のエネルギー準位は、分子全体のエネルギー状態を決定します。電子は通常、エネルギーが最も低い軌道から順に配置されます。
- 電子配置: 分子内の電子は、パウリの排他原理およびフントの規則に従って配置されます。これにより、分子の安定性や反応性が決まります。
- 化学結合: 分子軌道理論により、化学結合の形成過程が定量的に説明されます。結合軌道に電子が配置されることで、結合エネルギーが低下し、分子が安定化します。
1.5 分子軌道の視覚化
分子軌道を視覚化することで、分子内の電子の分布や結合の性質を直感的に理解することができます。視覚化ツールとしては、Py3DmolやAvogadroなどのソフトウェアが利用され、これにより分子軌道の形状やエネルギー準位を具体的に把握することが可能です。今回はPy3Dmolを使用します。
2. HOMOとLUMOの性質
2.1 HOMO、LUMOの性質
HOMO(Highest Occupied Molecular Orbital、最高被占分子軌道)とLUMO(Lowest Unoccupied Molecular Orbital、最低空分子軌道)は、分子軌道理論において特に重要な役割を果たすフロンティア軌道です。これらの軌道は、分子の反応性や電子特性に深く関与します[2]。
- HOMOの性質:
- HOMOは、分子内で最も高いエネルギーを持つ電子が存在する軌道です。HOMOの電子は、外部からの刺激に対して反応しやすく、特に酸化反応において重要です。HOMOのエネルギーが高いほど、分子は電子を供与しやすくなります。
- 例えば、電子供与性の高い置換基を持つ分子では、HOMOのエネルギーが上昇し、反応性が増加します。
- LUMOの性質:
- LUMOは、分子内で最も低いエネルギーを持つ空の軌道です。LUMOは、外部から電子を受け取る能力を持ち、特に還元反応において重要です。LUMOのエネルギーが低いほど、分子は電子を受容しやすくなります。
- 例えば、電子求引性の高い置換基を持つ分子では、LUMOのエネルギーが低下し、反応性が増加します。
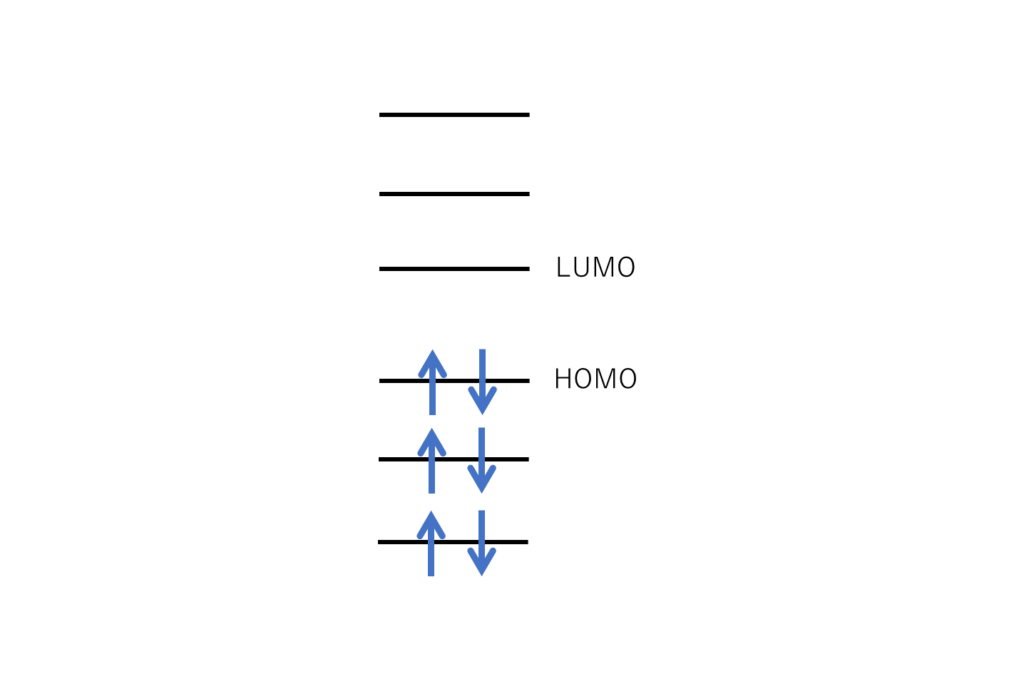
2.2 フロンティア軌道と反応性
フロンティア軌道理論は、HOMOとLUMOのエネルギーレベルが分子の化学反応性に与える影響を説明する理論です。特に、HOMO-LUMOギャップ(エネルギー差)は、分子の安定性や反応性を左右します。
- HOMO-LUMOギャップ:
- HOMOとLUMOのエネルギー差が大きいほど、分子は安定し、化学反応に対して鈍感になります。一方、ギャップが小さいほど、分子は反応性が高くなります。
- 有機半導体において、HOMO-LUMOギャップは電子輸送特性や光電変換効率に直接影響します。例えば、ポリチオフェンやポリパラフェニレンビニレン(PPV)などの有機半導体材料では、ギャップの調整によりデバイスの性能を最適化できます。
- フロンティア軌道と化学反応:
- フロンティア軌道理論は、電子の供与と受容を通じて化学反応を説明します。例えば、Diels-Alder反応では、ジエンのHOMOとジエノフィルのLUMOの重なりが反応を駆動します。このように、反応性はフロンティア軌道のエネルギー配置に依存します。
- 有機半導体におけるフロンティア軌道:
- 有機半導体は、エレクトロニクスやフォトニクスの分野で重要な材料です。これらの材料のHOMOとLUMOのエネルギーレベルは、電荷輸送特性や光吸収特性に影響を与えます。
- 例えば、ポリチオフェンでは、HOMOのエネルギーを高く保ちつつ、LUMOのエネルギーを低くすることで、高効率の太陽電池や有機トランジスタが実現できます。これにより、デバイスの性能が向上し、エネルギー変換効率が高まります。
2.3 具体例:ベンゼンと置換基
以下に、ベンゼン分子とその置換基におけるHOMOとLUMOのエネルギー準位の変化を示します。
- ベンゼンのHOMOとLUMO:
- ベンゼン分子のHOMOとLUMOのエネルギー準位は、それぞれ-6.69 eVと-0.09 eVです。
- 置換基が導入されると、これらのエネルギー準位は変化します。
- 置換基の影響:
- 電子供与性の置換基(例:NH2基)は、HOMOのエネルギーを上昇させ、LUMOのエネルギーをわずかに変化させます。これは、置換基のp軌道性の高い被占軌道に収まっている非共有電子対と縮退するベンゼン環のHOMOの内の一つが反結合性軌道相互作用を起こすことが理由に考えられます。
- 電子求引性の置換基(例:NO2基)は、HOMOのエネルギーを低下させ、LUMOのエネルギーを顕著に低下させます。これは、置換基のp軌道性の高い被占軌道に収まっている非共有電子対と縮退するベンゼン環のHOMOの内の一つが結合性軌道相互作用を起こすことが理由に考えられます。
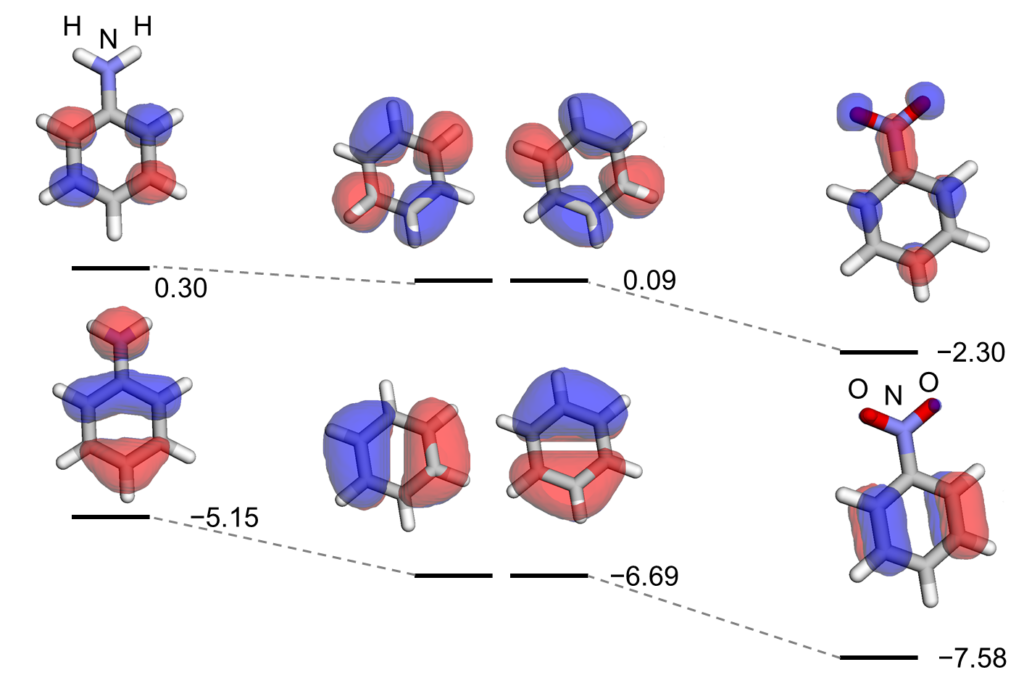
3. PySCFによる分子軌道可視化手順
これまでの章で、分子軌道の基本概念やHOMOとLUMOの性質について学びました。ここでは、実際にPySCFを用いて分子軌道を計算し、その結果を可視化する方法を具体的に解説します。以下の手順に従って、ベンゼン分子を例に分子軌道を計算・可視化していきましょう。
今回はGoogle Colabを使用しています。
Google Colabは、ブラウザベースのPythonプログラミング環境であり、PySCFを使用するのに適したプラットフォームの一つです。Google Colabを使用することで、ローカル環境のセットアップなしにPySCFの計算をすぐに開始することができます。
Google colabのリンク:https://colab.research.google.com/?hl=ja
3.1 PySCFのインストール
PySCFを使用するために必要なパッケージをインストールします。Google Colabを使用している場合は、以下のコマンドを実行してください[3-8]。
!pip install pyscf
!pip install numpy
!pip install geometric
!pip install py3Dmol3.2 PySCFの基本設定
まずは、PySCFを使ってベンゼン分子の量子化学計算を行うための基本設定を行います。
from pyscf import gto, dft, lib
from pyscf.geomopt import geometric_solver
# ベンゼン分子の定義
mol = gto.M( atom='''
C -1.6506218707 -0.3639250453 -0.1642271309
C -1.4278287046 -1.7356986384 -0.0032249128
C -2.5123496441 -2.6159337317 0.0762847300
C -3.8196637673 -2.1243951231 -0.0052067680
C -4.0424569309 -0.7526215232 -0.1662088378
C -2.9579359796 0.1276134478 -0.2457195582
H -3.1302950692 1.1888560352 -0.3702759564
H -0.8116059456 0.3170495820 -0.2257382130
H -0.4164537219 -2.1159666701 0.0598199268
H -2.3399905562 -3.6771764633 0.2008399154
H -4.6586797260 -2.8053696634 0.0563047720
H -5.0538319298 -0.3723534259 -0.2292529483 ''', basis='6-31G(d)', spin=0, # スピンを指定 charge=0, # 電荷を指定 verbose=0
)
# DFT計算のセットアップ
mf = dft.RKS(mol)
mf.chkfile = 'checkpoint_file.chk'
mf.xc = 'B3LYP'
mf.max_cycle = 150 # SCF計算の最大反復回数
# 構造最適化
geometric_solver.optimize(mf)このコードでは、ベンゼン分子の原子座標を定義し、DFT(Density Functional Theory)計算をセットアップしています。ここでは、B3LYPという一般的な汎関数を使用し、計算の最大反復回数を150回に設定しています。構造最適化はgeometric_solverを使用して行われます。
3.3 HOMOとLUMOの計算と確認
次に、構造最適化後のベンゼン分子に対してSCF(Self-Consistent Field)計算を再実行し、HOMO(最高被占分子軌道)およびLUMO(最低空分子軌道)のエネルギーを計算します。
# 最適化後のジオメトリーを取得
final_geometry = mol.atom_coords(unit="ANG")
# SCF計算の再実行
mf = dft.RKS(mol)
mf.chkfile = 'checkpoint_file.chk'
mf.xc = 'B3LYP'
mf.kernel()
# 計算結果の検証
if mf.mo_occ is not None and len(mf.mo_occ) > 0: print("SCF calculation completed successfully.") print(f"Number of occupied orbitals: {sum(mf.mo_occ > 0)}")
else: raise ValueError("SCF calculation failed or mo_occ is not properly assigned.")
# HOMOおよびLUMOのインデックスを取得
homo_index = (mf.mo_occ > 0).nonzero()[0][-1]
lumo_index = (mf.mo_occ == 0).nonzero()[0][0]このコードでは、最適化後の分子構造を取得し、SCF計算を再実行しています。計算結果からHOMOとLUMOのインデックスを取得し、それぞれのエネルギーレベルを確認します。
3.4 CUBEファイルの生成と可視化
計算結果を基に、指定した分子軌道のCUBEファイルを生成し、可視化ツールを用いて分子軌道を視覚的に表示します。
from pyscf import tools
import py3Dmol
from IPython.display import display, HTML
import ipywidgets as widgets
# 指定した軌道のCUBEファイルを生成する関数
def generate_cube_file(orbital_index, filename): tools.cubegen.orbital(mol, filename, mf.mo_coeff[:, orbital_index])
# 可視化する軌道のリストを作成
orbitals = { "LUMO+3": lumo_index + 3, "LUMO+2": lumo_index + 2, "LUMO+1": lumo_index + 1, "LUMO": lumo_index, "HOMO": homo_index, "HOMO-1": homo_index - 1, "HOMO-2": homo_index - 2, "HOMO-3": homo_index - 3
}
# エネルギーをeVに変換
hartree_to_ev = 27.2114
orbital_energies = {name: mf.mo_energy[idx] * hartree_to_ev for name, idx in orbitals.items()}
# ウィジェットの作成
dropdown = widgets.Dropdown( options=[(f"{name} ({orbital_energies[name]:.6f} eV)", name) for name in orbitals.keys()], description='Orbital:', disabled=False,
)
output = widgets.Output()
# 可視化関数
def visualize_orbital(orbital_name): orbital_index = orbitals[orbital_name] cube_filename = f'{orbital_name}.cube' generate_cube_file(orbital_index, cube_filename) view = py3Dmol.view(width=800, height=600) with open(cube_filename) as f: cube = f.read() view.addVolumetricData(cube, 'cube', {'isoval': 0.02, 'color': 'red', 'opacity': 0.75}) view.addVolumetricData(cube, 'cube', {'isoval': -0.02, 'color': 'blue', 'opacity': 0.75}) # 最適化後の分子形状を追加 xyz = "\\\\n".join([f"{mol.atom_symbol(i)} {final_geometry[i][0]:.6f} {final_geometry[i][1]:.6f} {final_geometry[i][2]:.6f}" for i in range(mol.natm)]) xyz = f"{mol.natm}\\\\n\\\\n" + xyz # xyzフォーマットのヘッダー追加 view.addModel(xyz, 'xyz') view.setStyle({'stick': {}}) view.zoomTo() # エネルギーのラベルを追加 orbital_energy = orbital_energies[orbital_name] view.addLabel(f'{orbital_name}: {orbital_energy:.6f} eV', {'fontSize': 12, 'fontColor': 'black', 'backgroundColor': 'white', 'backgroundOpacity': 0.8, 'showBackground': True, 'position': {'x': 0, 'y': 0, 'z': 0}}) with output: output.clear_output(wait=True) display(HTML(view._make_html()))
# ドロップダウンの変更イベントに可視化関数をバインド
def on_dropdown_change(change): with output: output.clear_output(wait=True) visualize_orbital(change['new'])
dropdown.observe(on_dropdown_change, names='value')
# インターフェースの表示
display(dropdown)
display(output)
# 初期表示
visualize_orbital(list(orbitals.keys())[0])このコードでは、HOMOおよびLUMOのCUBEファイルを生成し、py3Dmolを用いて分子軌道を可視化しています。visualize_orbital関数を使用して、CUBEファイルを読み込み、軌道の形状を3Dで表示します。また、最適化後の分子形状も表示されるようにしています。
出力が上手くいけば以下のような画像が得られます。
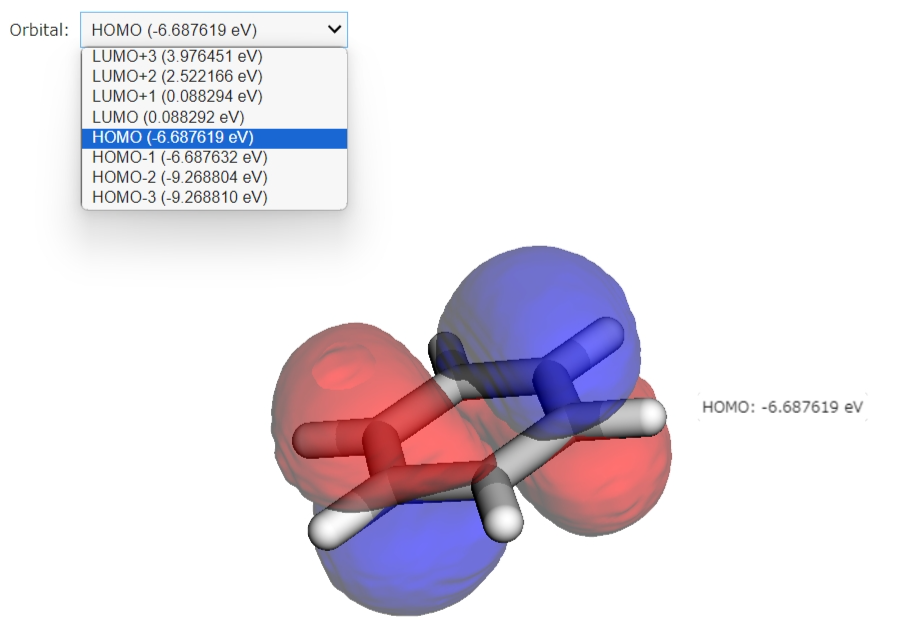
3.5 各軌道のエネルギーの見方
最後に、各軌道のエネルギーを確認します。エネルギーはハートリー単位から電子ボルト(eV)に変換され、可視化時に表示されます。
# エネルギーをeVに変換
hartree_to_ev = 27.2114
homo_energy = mf.mo_energy[homo_index] * hartree_to_ev
lumo_energy = mf.mo_energy[lumo_index] * hartree_to_ev
print(f'HOMO Energy: {homo_energy:.6f} eV')
print(f'LUMO Energy: {lumo_energy:.6f} eV')終わりに
PySCFを用いた分子軌道の計算と可視化の手順について説明しました。PySCFは高い拡張性とカスタマイズ性を持つため、様々な分子系の解析に利用することができます。是非、この記事を参考にして、PySCFを使った量子化学計算を実践してみてください。
参考文献
[1] PySCF 2.4 公式サイト, https://pyscf.org/, (参照2024-06-24)[2] 西長亨、本田康 「有機化学者のための量子化学計算入門 Gaussianの基本と有効利用のヒント」 P. 110-113[3] Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Koval, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Scheurer, H. F. Schurkus, J. E. T. Smith, C. Sun, S.-N. Sun, S. Upadhyay, L. K. Wagner, X. Wang, A. White, J. Daniel Whitfield, M. J. Williamson, S. Wouters, J. Yang, J. M. Yu, T. Zhu, T. C. Berkelbach, S. Sharma, A. Yu. Sokolov, and G. K.-L. Chan, Recent developments in the PySCF program package, J. Chem. Phys. 153, 024109 (2020)[4] Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. McClain, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: the Python-based simulations of chemistry framework, WIREs Comput. Mol. Sci. 8, e1340 (2018)[5]Ulf Ekström, Lucas Visscher, Radovan Bast, Andreas J. Thorvaldsen, and Kenneth Ruud, Arbitrary-order density functional response theory from automatic differentiation, J. Chem. Theory Comput. 6, 1971 (2010).[6] Susi Lehtola, Conrad Steigemann, Micael J.T. Oliveira, and Miguel A.L. Marques, Recent developments in libxc — A comprehensive library of functionals for density functional theory, SoftwareX 7, 1 (2018).[7] L.-P. Wang, C.C. Song, “Geometry optimization made simple with translation and rotation coordinates”, J. Chem, Phys. 144, 214108 (2016).[8] py3Dmol in Jupyter, https://birdlet.github.io/2019/10/02/py3dmol_example/ (参照2024-06-24)
Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

