

本記事はin silico screeningにおけるタンパク質準備について書かれた記事です。OpenMMを使って、タンパク質のエネルギー最小化とオープンソース版のPymolを使ったgrid boxの作成方法を記載しています。企業の方でも使える内容ですので、ぜひご覧ください!
【この記事のまとめ】
in silico創薬に取り組む研究者や学生に向け、OpenMMを用いたタンパク質のエネルギー最小化と、PyMOLを活用したドッキングシミュレーション用グリッドボックス(Grid box)の作成方法を解説した実践ガイドです。精度の高いシミュレーションに不可欠な「構造の最適化」と「探索領域の視覚化」を、具体的なコードと共に再現可能な形で提示しています。
- OpenMMによる構造最適化: Python APIを活用し、タンパク質への水素原子追加からエネルギー最小化までを数行のコードで完結させる手順を公開。
- PyMOLでのGrid box可視化: カスタムPythonスクリプト(grid.py)を用い、標的部位の中心座標(Center of mass)とボックスサイズを自動算出し、画面上に描画する方法を詳説。
- 環境構築の自動化:
condaを用いたOpenMMおよびオープンソース版PyMOLの効率的なインストール手順を網羅し、企業・アカデミアを問わず導入をサポート。
この記事を読むことで、手作業でのミスが起きやすい座標設定を自動化し、ドッキングシミュレーションの基盤となるタンパク質準備を正確かつ迅速に行えるようになります。
Mac M1, Sequoia 15.3

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


OpenMMとは
OpenMM(オープンエムエム)は、Python や C++ から呼び出せる GPU対応の分子動力学(MD)シミュレーション・ライブラリです。CUDA または OpenCL を活用でき、数万原子規模の系でも実用的な速度でカスタム計算を行えます。
主な特長:
- Python API を介して、
ForceFieldやSimulation、カスタム積分器などを柔軟に組み立て可能です。。 - GPU 有効化 により、CUDA/OpenCL ベースの高速化が進んでおり、複数GPUでの並列実行もサポートされます。
- 構造最適化(minimize) やサンプリング、自由エネルギー計算などの基本機能を備えています。
ライセンスは、API や CPU リファレンス部分が MIT ライセンス(商用・改変可)で公開されており、CUDA/OpenCL プラットフォーム部分は LGPL ライセンス です。
総じて、OpenMM は、柔軟なカスタマイズ性とGPUによる高性能を両立し、研究用途から創薬シミュレーションまで幅広く利用できる強力なMDツールキットです。
ここではOpenMMを使って、エネルギーを最小化してみましょう!
過去記事ではGROMACSでもエネルギー最小化をしましたが、OpenMMの方がシンプルにエネルギーを最小化できます。
環境構築
可視化ツールであるPymol(オープンソース版)と共に、OpenMMの環境構築をしましょう!
conda create -n protein_preparation python=3.10 -y
conda activate protein_preparation
conda install -c conda-forge pymol-open-source
conda install -c conda-forge openmm
pymol
詳細コード解説
仮想環境の作成と起動
conda create -n protein_preparation python=3.10 -y
- 仮想環境
protein_preparationをPython 3.10で作成 y:確認なしで自動的にインストールを進める
conda activate protein_preparation
- 作成した仮想環境を有効化
- これ以降の操作はこの環境内で行われます
PyMOL オープンソース版のインストール
conda install -c conda-forge pymol-open-source
- conda-forge チャンネルからPyMOLオープンソース版をインストール
- 商用ライセンス不要で、拡張機能やスクリプト利用も可
注意点:
- Schrödinger版とは異なり、GUIの見た目や機能が少しシンプルです
- Macでは起動時に
XQuartz(X11)が必要になることがあります
OpenMMのインストール
conda install -c conda-forge openmm
- 分子力学シミュレーションエンジン OpenMM をインストール
- Pythonから呼び出してエネルギー最小化やMDを行うことができます
PyMOLの起動
pymol
- PyMOLを起動します(GUIが立ち上がります)
- PyMOLの下部ターミナルでPythonコードやスクリプトを実行できます
Pymolを使ったタンパク質の準備
まずPymolが開かれると、以下の画面になっていると思います。左上のFIleからタンパク質をロードしてください。
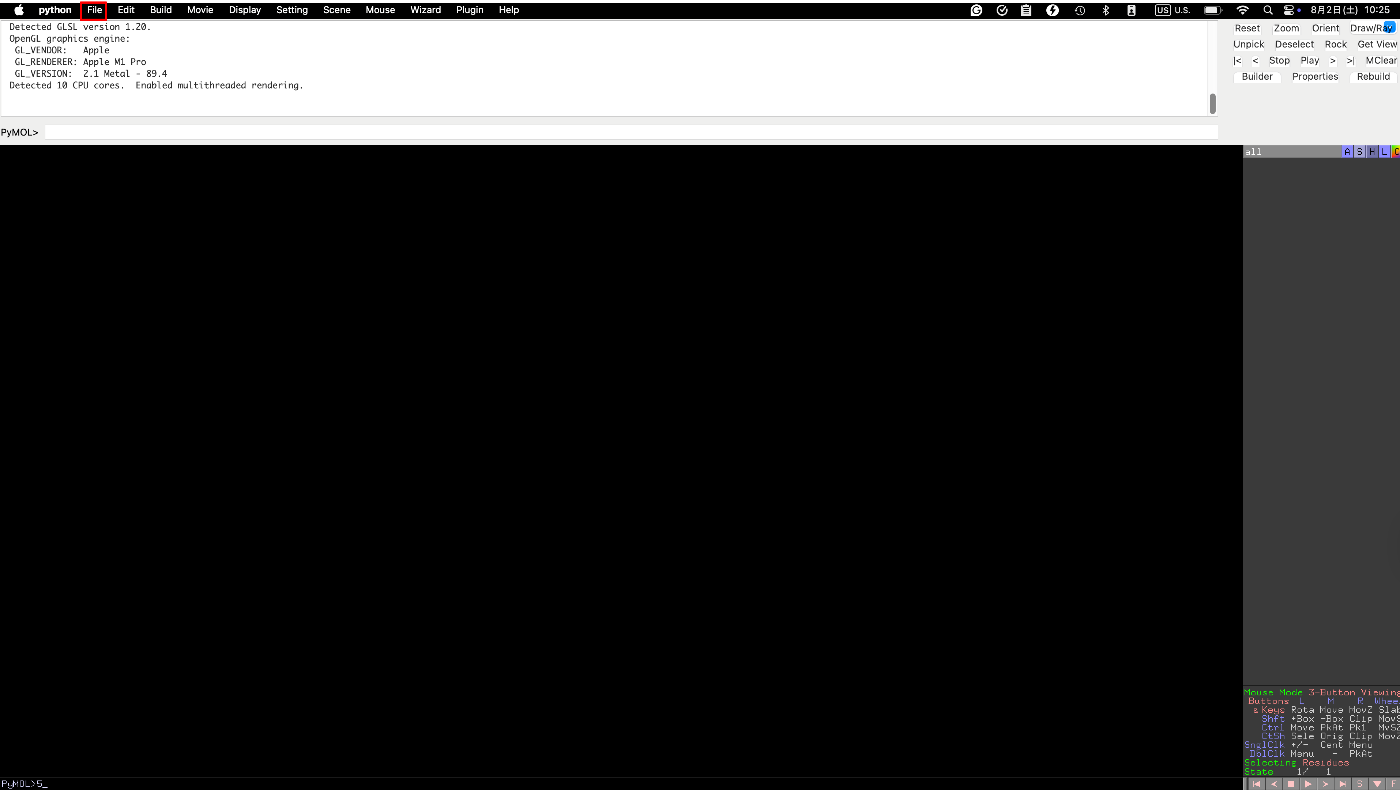
まずは水分子を消しましょう。
次に余分なイオンとリガンドを消していきます。
上タブのDisplay→Sequenceを押して、配列を出しましょう。
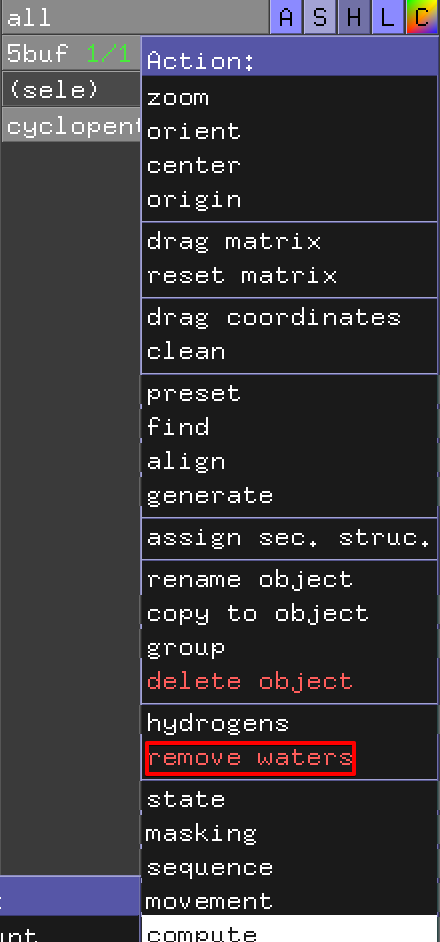
イオンとリガンドを選択し、右側のremove atomsから消してください。

左上のFile→Export Moleculesを押し、5buf_no_water_ligand.pdb として保存してください。
OpenMMを使ったエネルギー最小化
続いて、OpenMMを使って、エネルギーを最小化していきます・
全コードはこちら
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
# 1. PDB読み込み
pdb = PDBFile('/path/to/5buf_no_water_ligand.pdb')
# 2. ForceField定義
forcefield = ForceField('amber99sb.xml', 'tip3p.xml')
# 3. Modellerを使って水素原子を追加
modeller = Modeller(pdb.topology, pdb.positions)
modeller.addHydrogens(forcefield)
# 4. システム構築
system = forcefield.createSystem(
modeller.topology,
nonbondedMethod=PME,
nonbondedCutoff=1*nanometer,
constraints=HBonds
)
# 5. 積分器の定義
integrator = LangevinIntegrator(300*kelvin, 1/picosecond, 0.002*picoseconds)
# 6. シミュレーション準備
simulation = Simulation(modeller.topology, system, integrator)
# 7. 位置を設定
simulation.context.setPositions(modeller.positions)
# 8. エネルギー最小化を実行
print("エネルギー最小化を実行中...")
simulation.minimizeEnergy()
# もっとしたい場合はこちら
#simulation.minimizeEnergy(
# tolerance=10 * kilojoule_per_mole / nanometer,
# maxIterations=1000
#)
print("エネルギー最小化完了!")
# 9. 最小化後の構造を取得
positions = simulation.context.getState(getPositions=True).getPositions()
# 10. PDBファイルに出力
with open('minimized.pdb', 'w') as output_file:
PDBFile.writeFile(modeller.topology, positions, output_file)
print("最小化された構造を 'minimized.pdb' に保存しました。")
詳細コード
必要なモジュールのインポート
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
openmm.app:構造データの読み込み・操作、シミュレーション設定に使います。openmm:シミュレーションエンジンそのものです。openmm.unit:温度や長さなどに単位(K, nmなど)を付けるために使います。sys.stdout:ログやメッセージの表示先に使うこともあります(今回は未使用でもOK)。
PDBファイルの読み込み
pdb = PDBFile('/path/to/5buf_no_water_ligand.pdb')
PDBFileで構造ファイル(.pdb)を読み込みます。- ここでは水やリガンドが除かれたPDBを使用。
ForceFieldの定義
forcefield = ForceField('amber99sb.xml', 'tip3p.xml')
amber99sb.xml:タンパク質に使われる力場。tip3p.xml:水分子に対する力場(今回はまだ水は加えていませんが、後で使う可能性あり)。
Modellerを使って水素原子を追加
modeller = Modeller(pdb.topology, pdb.positions)
modeller.addHydrogens(forcefield)
- PDB構造には通常水素が含まれていないため、自動で水素を追加します。
Modellerオブジェクトで、構造の修正を行えます。
システムの構築
system = forcefield.createSystem(
modeller.topology,
nonbondedMethod=PME,
nonbondedCutoff=1*nanometer,
constraints=HBonds
)
- 力場とトポロジー情報から力の計算モデル(System)を生成。
PME:長距離の電荷相互作用を正確に扱う方法。HBonds:水素結合のみを固定(計算の高速化と安定化)。
積分器(Integrator)の設定
integrator = LangevinIntegrator(300*kelvin, 1/picosecond, 0.002*picoseconds)
- 温度を制御しながら時間発展を行うLangevin Dynamicsを使用。
- 300Kで、1/ps の摩擦係数、2 fs のタイムステップ。
Simulationオブジェクトの作成
simulation = Simulation(modeller.topology, system, integrator)
- 上記で作成したトポロジー・力場・積分器を使って、シミュレーション本体を準備。
初期構造の設定
simulation.context.setPositions(modeller.positions)
- 初期の原子座標をシミュレーションに反映させます。
エネルギー最小化を実行
print("エネルギー最小化を実行中...")
simulation.minimizeEnergy()
#もっと厳密にエネルギー最小化をしたい場合はこちら
#simulation.minimizeEnergy(
# tolerance=10 * kilojoule_per_mole / nanometer,
# maxIterations=1000
#)
print("エネルギー最小化完了!")
- 原子間の衝突や不自然な構造を修正(エネルギー最小化)します。
- 最小化によって、より現実的な構造へと近づけます。
パラメータを調整することで、さらに厳密にエネルギー最小化を行えます。
maxIterations の値を変更してみてください。
最小化後の構造を取得
positions = simulation.context.getState(getPositions=True).getPositions()
- 最小化された座標を取得します。
- この座標を使って新しいPDBファイルを書き出せます。
最小化構造のPDBファイル出力
with open('minimized.pdb', 'w') as output_file:
PDBFile.writeFile(modeller.topology, positions, output_file)
print("最小化された構造を 'minimized.pdb' に保存しました。")
writeFile()でPDB形式として保存します。- これにより、水素付き&最小化済みの構造が
minimized.pdbに出力されます。
PymolでのGridBoxの作り方
ketanne さんが記事にまとめてくれていました。この通りにGridBoxを作ります。
PyMOLでDocking Simulationできるのか -gridboxを作る編-
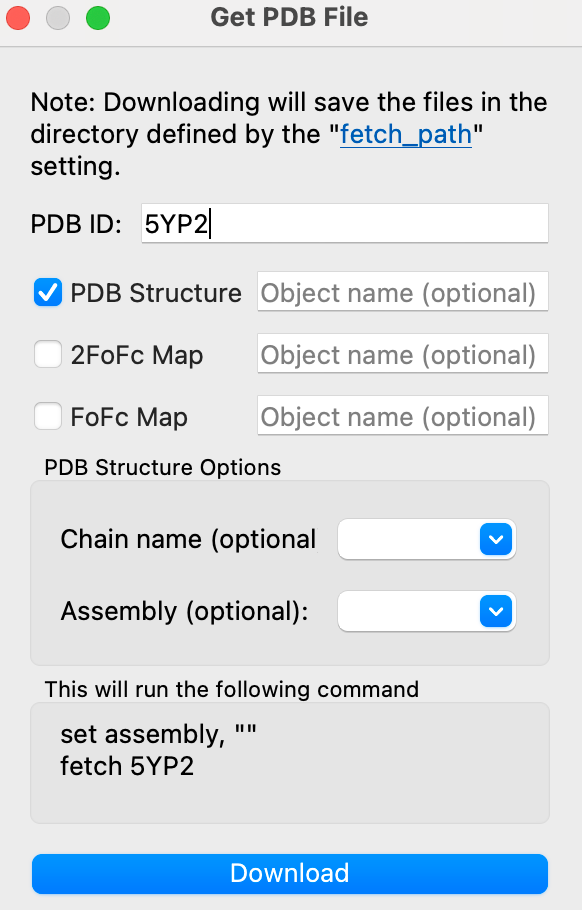
今回はわかりやすいようにDPP4(PDB: 5YP2)を用いて、すでに結合しているinhibitor周辺のGrid boxを見ていきます。
左上にあるFile→Get PDB File→PDB IDに5YP2を入れて、Downloadを押してください。
その後、上タブのDisplay→Sequenceを押して、配列を出し、水分子を消してください。
次にリガンドを一つのみ(今回は8YC)残し、残りは消してください。
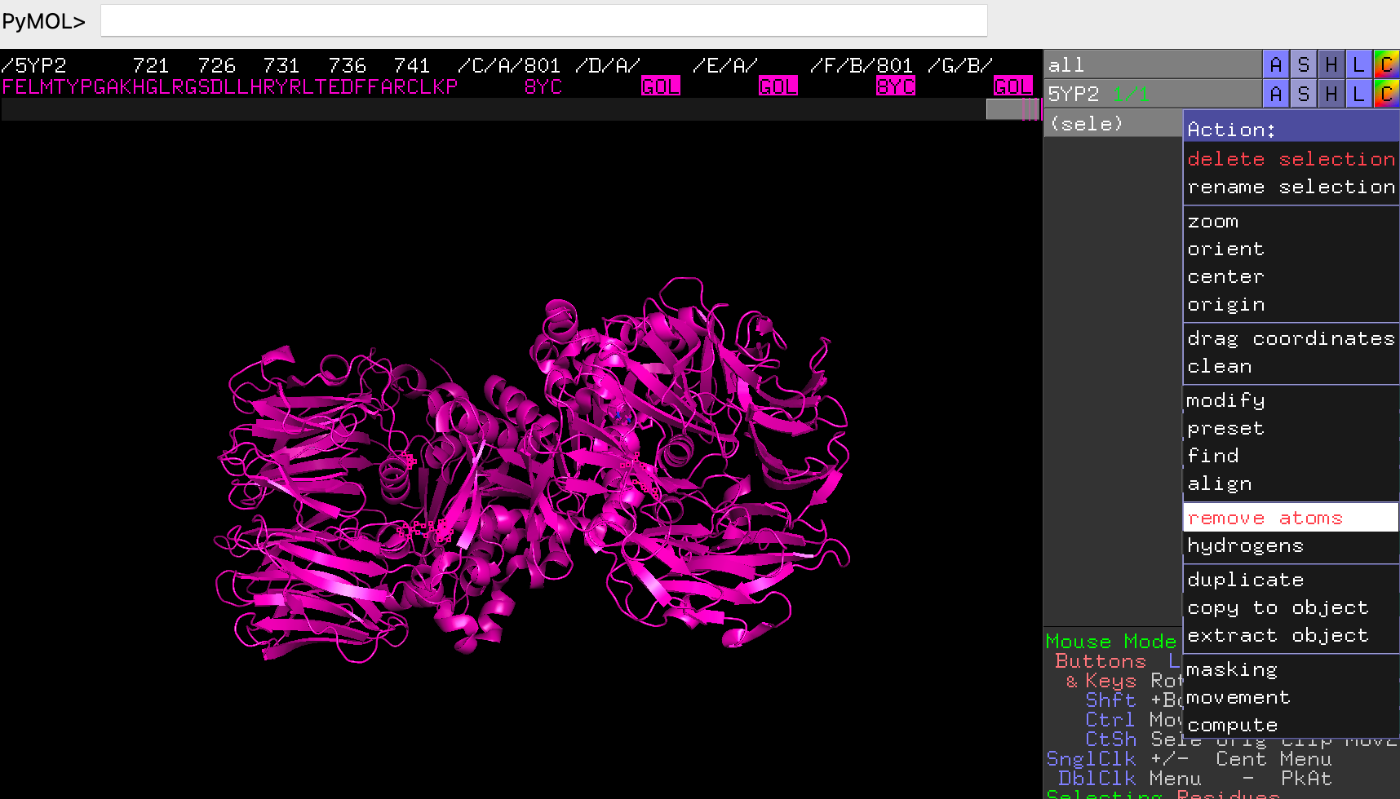
以下のPythonのスクリプトをgrid.py として、保存してください。
grid.py
from pymol import cmd
from pymol.cgo import *
import itertools
from random import randint
def search_space(selection="enabled and organic", padding=4.0):
([minX, minY, minZ], [maxX, maxY, maxZ]) = cmd.get_extent(selection)
print("Box dimensions (%.2f, %.2f, %.2f)" % (maxX - minX, maxY - minY, maxZ - minZ))
center_of_mass = [(minX + maxX) / 2, (minY + maxY) / 2, (minZ + maxZ) / 2]
print("Center of mass (%.2f, %.2f, %.2f)" % tuple(center_of_mass))
minX = minX - float(padding)
minY = minY - float(padding)
minZ = minZ - float(padding)
maxX = maxX + float(padding)
maxY = maxY + float(padding)
maxZ = maxZ + float(padding)
size_x = maxX - minX
size_y = maxY - minY
size_z = maxZ - minZ
center_x = (minX + maxX) / 2
center_y = (minY + maxY) / 2
center_z = (minZ + maxZ) / 2
return size_x, size_y, size_z, center_x, center_y, center_z
print("\nSmina Options:")
print("--center_x %.3f," % center_x, "--center_y %.3f," % center_y, "--center_z %.3f," % center_z,
"--size_x %.3f," % size_x, "--size_y %.3f," % size_y, "--size_z %.3f," % size_z)
cmd.extend("search_space", search_space)
# draw gridbox
def grid(selection="enabled and organic", padding=4.0, lw=1.5, r=0.5, g=0.5, b=0.8):
([minX, minY, minZ], [maxX, maxY, maxZ]) = cmd.get_extent(selection)
print("Box dimensions (%.2f, %.2f, %.2f)" % (maxX - minX, maxY - minY, maxZ - minZ))
center_of_mass = [(minX + maxX) / 2, (minY + maxY) / 2, (minZ + maxZ) / 2]
print("Center of mass (%.2f, %.2f, %.2f)" % tuple(center_of_mass))
minX = minX - float(padding)
minY = minY - float(padding)
minZ = minZ - float(padding)
maxX = maxX + float(padding)
maxY = maxY + float(padding)
maxZ = maxZ + float(padding)
box = [
LINEWIDTH, float(lw),
BEGIN, LINES,
COLOR, float(r), float(g), float(b),
VERTEX, minX, minY, minZ, VERTEX, maxX, minY, minZ,
VERTEX, minX, maxY, minZ, VERTEX, maxX, maxY, minZ,
VERTEX, minX, minY, maxZ, VERTEX, maxX, minY, maxZ,
VERTEX, minX, maxY, maxZ, VERTEX, maxX, maxY, maxZ,
VERTEX, minX, minY, minZ, VERTEX, minX, maxY, minZ,
VERTEX, maxX, minY, minZ, VERTEX, maxX, maxY, minZ,
VERTEX, minX, minY, maxZ, VERTEX, minX, maxY, maxZ,
VERTEX, maxX, minY, maxZ, VERTEX, maxX, maxY, maxZ,
VERTEX, minX, minY, minZ, VERTEX, minX, minY, maxZ,
VERTEX, maxX, minY, minZ, VERTEX, maxX, minY, maxZ,
VERTEX, minX, maxY, minZ, VERTEX, minX, maxY, maxZ,
VERTEX, maxX, maxY, minZ, VERTEX, maxX, maxY, maxZ,
END
]
boxName = "box_" + str(randint(0, 10000))
while boxName in cmd.get_names():
boxName = "box_" + str(randint(0, 10000))
cmd.load_cgo(box, boxName)
return boxName
cmd.extend("grid", grid)
このスクリプトは、PyMOLで指定した分子(デフォルトではリガンド)の空間範囲(search space)を計算し、その範囲にグリッドボックスを描画するものです。AutoDock VinaやSminaのドッキングに必要なcenter座標とsizeも取得できます。
詳細コード
必要なモジュールのインポート
from pymol import cmd
from pymol.cgo import *
import itertools
from random import randint
cmd:PyMOLの内部コマンドにアクセスするためのインターフェースcgo:Custom Graphics Object(線などの3Dオブジェクトを描画)randint:重複のないボックス名を作成するための乱数生成
search_space関数:ドッキング空間の座標情報を取得
def search_space(selection="enabled and organic", padding=4.0):
([minX, minY, minZ], [maxX, maxY, maxZ]) = cmd.get_extent(selection)
print("Box dimensions (%.2f, %.2f, %.2f)" % (maxX - minX, maxY - minY, maxZ - minZ))
center_of_mass = [(minX + maxX) / 2, (minY + maxY) / 2, (minZ + maxZ) / 2]
print("Center of mass (%.2f, %.2f, %.2f)" % tuple(center_of_mass))
minX -= float(padding)
minY -= float(padding)
minZ -= float(padding)
maxX += float(padding)
maxY += float(padding)
maxZ += float(padding)
size_x = maxX - minX
size_y = maxY - minY
size_z = maxZ - minZ
center_x = (minX + maxX) / 2
center_y = (minY + maxY) / 2
center_z = (minZ + maxZ) / 2
return size_x, size_y, size_z, center_x, center_y, center_z
cmd.get_extent(selection)で分子のXYZの範囲を取得padding値を加えて、グリッドボックスを少し大きく設定size_x, size_y, size_z:ボックスのサイズcenter_x, center_y, center_z:ボックスの中心(ドッキングに使う座標)
🔧 PyMOL上で search_space() を呼び出すと、コンソールに中心座標とサイズが出力されます。
PyMOLコマンドとして拡張
cmd.extend("search_space", search_space)
- PyMOL内で
search_spaceコマンドとして呼び出せるようにします。
grid関数:グリッドボックスの描画
def grid(selection="enabled and organic", padding=4.0, lw=1.5, r=0.5, g=0.5, b=0.8):
([minX, minY, minZ], [maxX, maxY, maxZ]) = cmd.get_extent(selection)
print("Box dimensions (%.2f, %.2f, %.2f)" % (maxX - minX, maxY - minY, maxZ - minZ))
center_of_mass = [(minX + maxX) / 2, (minY + maxY) / 2, (minZ + maxZ) / 2]
print("Center of mass (%.2f, %.2f, %.2f)" % tuple(center_of_mass))
minX -= float(padding)
minY -= float(padding)
minZ -= float(padding)
maxX += float(padding)
maxY += float(padding)
maxZ += float(padding)
box = [
LINEWIDTH, float(lw),
BEGIN, LINES,
COLOR, float(r), float(g), float(b),
VERTEX, minX, minY, minZ, VERTEX, maxX, minY, minZ,
VERTEX, minX, maxY, minZ, VERTEX, maxX, maxY, minZ,
VERTEX, minX, minY, maxZ, VERTEX, maxX, minY, maxZ,
VERTEX, minX, maxY, maxZ, VERTEX, maxX, maxY, maxZ,
VERTEX, minX, minY, minZ, VERTEX, minX, maxY, minZ,
VERTEX, maxX, minY, minZ, VERTEX, maxX, maxY, minZ,
VERTEX, minX, minY, maxZ, VERTEX, minX, maxY, maxZ,
VERTEX, maxX, minY, maxZ, VERTEX, maxX, maxY, maxZ,
VERTEX, minX, minY, minZ, VERTEX, minX, minY, maxZ,
VERTEX, maxX, minY, minZ, VERTEX, maxX, minY, maxZ,
VERTEX, minX, maxY, minZ, VERTEX, minX, maxY, maxZ,
VERTEX, maxX, maxY, minZ, VERTEX, maxX, maxY, maxZ,
END
]
cmd.get_extent()で選択領域の空間を取得し、paddingを加えて拡張- PyMOLのCGO(Custom Graphics Object)で12本の辺からなる立方体(グリッドボックス)を描画
- 色(RGB)や線の太さ(
lw)も指定可能
グリッドオブジェクトの登録と表示
boxName = "box_" + str(randint(0, 10000))
while boxName in cmd.get_names():
boxName = "box_" + str(randint(0, 10000))
cmd.load_cgo(box, boxName)
return boxName
randintを使って一意なボックス名(例:box_4928)を生成cmd.load_cgo()でCGOオブジェクトをPyMOLに読み込む
PyMOLコマンドとして拡張
cmd.extend("grid", grid)
- PyMOL上で
gridコマンドを使えるようにします(例:grid ligand)
使い方まとめ
▶ リガンドのドッキング領域のサイズと中心を調べたい場合:
search_space ligand
ligandはオブジェクトや選択の名前- 結果はPyMOLのターミナルに表示されます
▶ グリッドボックスを表示したい場合:
grid ligand
- 選択された領域を囲むボックスがPyMOL上に描画されます
💡補足
padding=4.0を調整するとボックスサイズが変わりますselection="ligand"を"resn ATP"などに変えると特定の残基にも対応可能
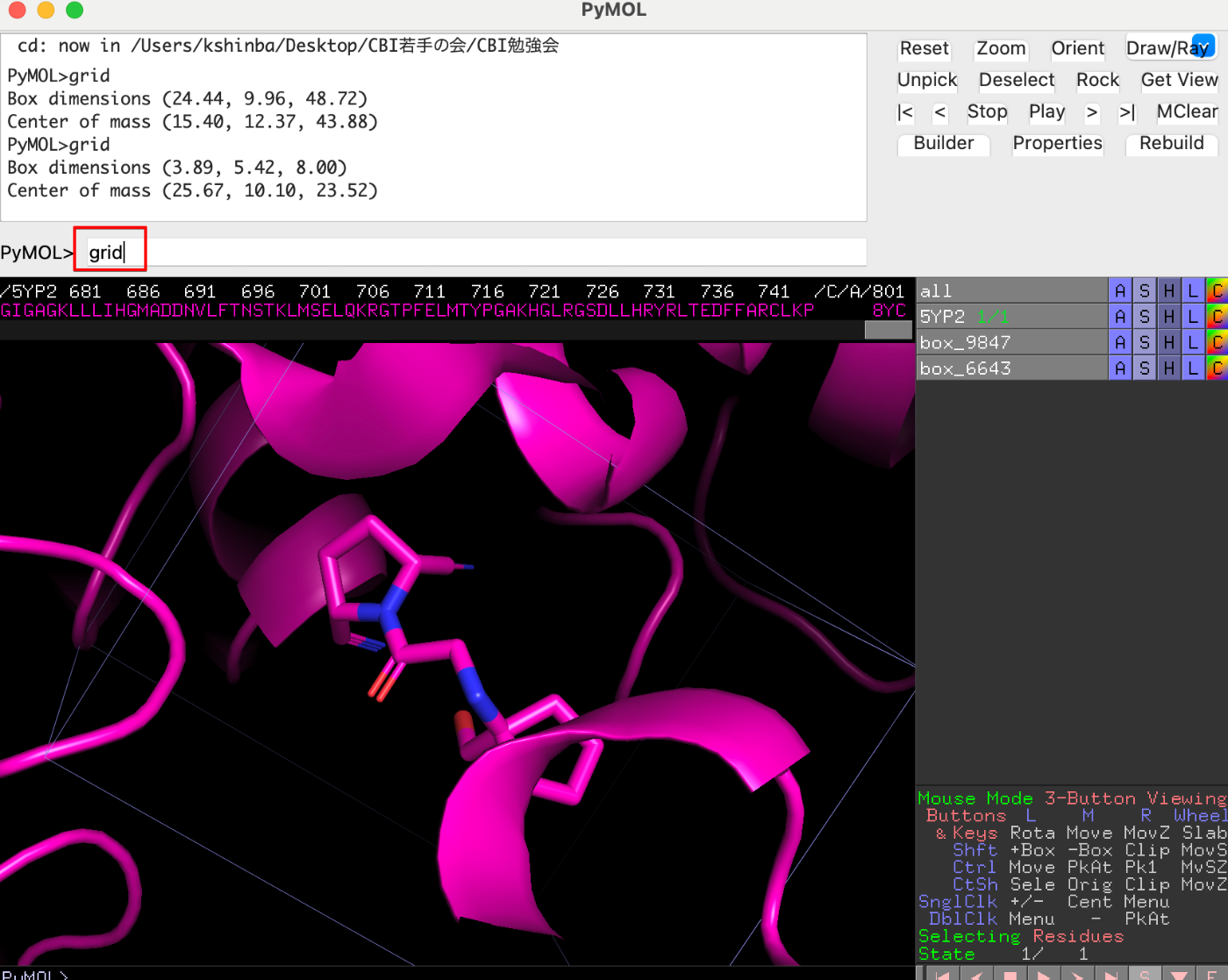
保存したファイルをFile→RunScript…でrunさせておきます。
gridとコマンドプロンプトに入力し、Enterを押すと、Box dimensionsなど出てきます!
このCenter of massを後のスクリーニングで記入すると良いでしょう。
最後に
過去記事では、UCFS Chimeraでのタンパク質のエネルギー最小化とgrid boxの作成についても記載しています。Windowsかつアカデミアの方についてはこの記事に従った方が少し楽かもしれません。やっていることはどちらでも変わらないので、お好きな方でお試しください!
参考文献
License: MIT, LGPL
License: BSD-2-Clause
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

