

本記事は環状ペプチドのin silicoスクリーニングの前段階の環状ペプチドライブラリを構築する記事です。HELM表記を使って、簡単にライブラリを構築することができます。ぜひ挑戦してみてください!
【この記事のまとめ】
創薬研究やバイオインフォマティクスに従事する方に向けて、HELM形式を活用してin silicoスクリーニング用の「環状ペプチドライブラリ」を効率的に構築・可視化する方法を解説します。
- HELM形式による複雑な構造記述:非天然アミノ酸や環状構造を機械可読な形式で表現する標準フォーマット「HELM」の基礎と、Pythonでの変換手法を習得できます。
- 変異体生成の自動化:アラニンスキャニングやホモログスキャニングを実装し、リード配列から多様なライブラリを自動生成するワークフローを紹介します。
- 3D可視化からドッキング準備まで:RDKitやpy3Dmolを用いた3D構造の生成・確認、さらにAutoDock等のシミュレーションに不可欠なPDBQT形式への保存手順までを網羅しています。
この記事を読むことで、手間のかかる環状ペプチドのライブラリ作成を自動化し、より高度なin silico創薬シミュレーションへスムーズに移行できるようになります。
動作検証済み環境
Google Colab

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


ライブラリとは?
ペプチドライブラリは、さまざまな配列を持つペプチドの集合で、in silico創薬においては特定の標的タンパク質に対する結合能を評価するために使用されます。スクリーニングでは、各ペプチドの3D構造を予測し、ドッキングや分子動力学シミュレーションを用いて、結合エネルギーや安定性を比較します。多様な配列を効率的に評価するため、バッチ処理・自動化が重要です。
HELM形式とは?
HELM(Hierarchical Editing Language for Macromolecules)は、ペプチド、核酸、糖鎖などの複雑な生体高分子を機械可読な形式で表現するための標準フォーマットです。特にin silico創薬やバイオライブラリのデータ管理で重要な役割を担います。
特徴:
- モノマー単位で階層的に記述(アミノ酸やヌクレオチドが基本単位)
- 非天然アミノ酸・化学修飾・分岐構造も記述可能
- 構造の一貫性や相互運用性を保ちながら、データベースやツール間で利用可能
ペプチドの基本構文
PEPTIDE1{A.C.G.Y.R}$$$$PEPTIDE1: 分子の種類(ここではペプチド){}: アミノ酸配列(モノマーコード)$$$$: 接続や末端の情報がない場合の終端記号
非天然アミノ酸の表現
HELMでは、非天然アミノ酸もカスタムモノマーとして表現できます。モノマー辞書に登録すれば自由な名前で記述可能です。
PEPTIDE1{A.Dap.Cit.Nle}$$$$Dap: 2,3-ジアミノプロピオン酸Cit: シトルリンNle: ノルロイシン
これらはすべて非天然(standard amino acid ではない)ですが、辞書に定義しておけば扱えます。こちらを使った方法についてはまた別記事で書きます。今回は天然アミノ酸に絞ります。
環状ペプチドの記述
環状構造は末端をリンクで接続することで表現します。たとえば、1番目と5番目をペプチド結合で環化する場合:
PEPTIDE1{A.C.G.D.E}$$$V2.0$$$PEPTIDE1,1:R3-5:R2$$PEPTIDE1,1:R3-5:R2: モノマー1のR3とモノマー5のR2を結合(N-C末端環化など)
この記述により、主鎖環状ペプチド(例:シクロスポリンなど)も構造的に正確に表現できます。
アラニンスキャニング、ホモログスキャニングとは?
アラニンスキャニング(Alanine Scanning)
アラニンスキャニングは、タンパク質やペプチドの特定残基をアラニンに一つずつ置換し、その機能や結合能への影響を評価する手法です。アラニンは小さく中性なため、置換によって立体構造の変化を最小限に抑えつつ、その残基の役割を明らかにできます。in silicoでは、各変異体の結合エネルギー(ΔΔG)を計算することで、重要な結合残基(ホットスポット)を同定できます。M
ホモログスキャニング(Homologue Scanning)
ホモログスキャニングは、特定の残基をアミノ酸の中で物理化学的に近い他の残基に置換し、機能や安定性への影響を調べる方法です。たとえばリジンをアルギニンやヒスチジンに、フェニルアラニンをチロシンに変えるなど、類似性を保った変異を試みます。アラニンスキャニングよりもより自然に近い変異探索ができるため、機能改変や最適化設計に適しています。
どちらもin silicoでのスクリーニングにより、実験前の候補絞り込みやタンパク質改変戦略の立案に活用されます。
環境構築
今回のコードはGoogle Colabで実装しました!
こちらから実行してみてください。環境構築は不要です。
上のランタイム→すべてのセルを実行で実行できます
実装コード
全コードはこちら
!pip install rdkit py3Dmol
!pip install meeko
!pip install gemmi
import rdkit
from rdkit import Chem
from rdkit.Chem import AllChem
import py3Dmol
from typing import List
import re
# 必要なライブラリをインポートしています
# rdkit: 化学構造の操作や3D生成を行うためのライブラリ
# py3Dmol: 分子の3D構造をブラウザ上で可視化するためのライブラリ
# re: 正規表現を使って文字列処理を行うための標準ライブラリ
# --- Helper Functions (補助関数) ---
def convert_FASTA_to_HELM(seq: str, cyclic: bool = True) -> List[str]: """ FASTA形式の配列(例: 'ACDEF')をHELM形式(階層的編集言語)に変換する関数です。 cyclic=True の場合、先頭と末尾を結合して環状ペプチドにします(バックボーン環化)。 """ # 1. 配列の文字をドット区切りにします(例: H.M.T...) polymer = ".".join(list(seq)) # PEPTIDE1という名前のポリマーとして定義します helm = f"PEPTIDE1{{{polymer}}}" if cyclic: # 2. 環状化の処理 # N末端(R1)とC末端(R2)を結合させる記述を追加します n = len(seq) # 接続の構文: SourcePoly,TargetPoly,SourceAtom:Att-TargetAtom:Att connection = f"PEPTIDE1,PEPTIDE1,1:R1-{n}:R2" helm += f"${connection}$$$$" else: # 環状化しない場合は終了記号のみ追加 helm += "$$$$" return [helm]
def parse_helm_to_seq(helm: str) -> str: """HELM文字列からアミノ酸配列のみを抽出する関数です。""" # 正規表現を使って PEPTIDE1{...} の中身を取り出します match = re.search(r'PEPTIDE1\{(.*?)\}', helm) if match: # ドットを取り除いて文字列として返します(例: H.M.T -> HMT) return match.group(1).replace('.', '') return ""
def alanine_scanning(helm_str: str) -> List[str]: """ アラニンスキャニングを行う関数です。 ペプチドの各アミノ酸を1つずつアラニン(A)に置換した変異体を作成します。 特定のアミノ酸が活性に重要かどうかを調べるためによく使われます。 """ seq = parse_helm_to_seq(helm_str) variants = [] for i in range(len(seq)): # 元々がアラニンの場合はスキップ if seq[i] == 'A': continue # i番目のアミノ酸を'A'に置き換えた新しい配列を作成 new_seq = seq[:i] + 'A' + seq[i+1:] # HELM形式に変換してリストに追加 variants.append(convert_FASTA_to_HELM(new_seq)[0]) return variants
def homolog_scanning(helm_str: str) -> List[str]: """ ホモログスキャニングを行う関数です。 各アミノ酸を、性質が似ている別のアミノ酸(ホモログ)に置換した変異体を作成します。 """ seq = parse_helm_to_seq(helm_str) # 置換ルール(辞書): キーのアミノ酸を、値のリストにあるアミノ酸に置換します homologs = { 'H': ['K', 'R'], 'K': ['R', 'H'], 'R': ['K', 'H'], # 塩基性アミノ酸間での置換 'D': ['E'], 'E': ['D'], # 酸性アミノ酸間 'N': ['Q'], 'Q': ['N'], # アミド基を持つもの同士 'S': ['T'], 'T': ['S'], # 水酸基を持つもの同士 'V': ['I', 'L'], 'L': ['I', 'V'], 'I': ['L', 'V'], # 疎水性アミノ酸間 'F': ['Y'], 'Y': ['F'], # 芳香族アミノ酸間 'A': ['G'], 'G': ['A'], # 小さいアミノ酸間 'M': ['L'] } variants = [] for i in range(len(seq)): aa = seq[i] # そのアミノ酸に対する置換ルールが存在する場合 if aa in homologs: for sub in homologs[aa]: # 置換して新しい配列を作成 new_seq = seq[:i] + sub + seq[i+1:] variants.append(convert_FASTA_to_HELM(new_seq)[0]) return variants
def helm_to_3d_mol(helm_str: str): """ HELM文字列をRDKitの分子オブジェクト(3D構造込み)に変換する関数です。 RDKitを使って直鎖ペプチドを作成し、化学的に結合させて環状にします。 """ # 1. 配列情報を取得 seq = parse_helm_to_seq(helm_str) if not seq: return None # 2. 配列から直鎖状のペプチド分子を作成 mol = Chem.MolFromSequence(seq) if mol is None: return None # 3. 環状化(Head-to-Tail cyclization)の処理 # 分子を編集可能な状態(RWMol)にします rwmol = Chem.RWMol(mol) # N末端(アミノ基)とC末端(カルボキシ基)の原子を探します # SMARTS記法というパターンマッチングを使っています n_matches = rwmol.GetSubstructMatches(Chem.MolFromSmarts("[NH2,NH3]")) # N末端 c_matches = rwmol.GetSubstructMatches(Chem.MolFromSmarts("C(=O)[OH]")) # C末端 if not n_matches or not c_matches: print(f"Could not identify termini for {seq}") return None # N末端の窒素原子のインデックス n_atom_idx = n_matches[0][0] # C末端の炭素原子と、脱離するOH基の酸素原子を特定します c_term_indices = c_matches[-1] # 最後に見つかったものをC末端と仮定 c_atom_idx = -1 leaving_o_idx = -1 for idx in c_term_indices: atom = rwmol.GetAtomWithIdx(idx) if atom.GetSymbol() == 'C': # 炭素に結合している酸素(OH)を探す for bond in atom.GetBonds(): neighbor = bond.GetOtherAtom(atom) if neighbor.GetSymbol() == 'O' and bond.GetBondType() == Chem.BondType.SINGLE: c_atom_idx = idx leaving_o_idx = neighbor.GetIdx() break if c_atom_idx != -1: break # ペプチド結合の形成(脱水縮合のシミュレーション) if c_atom_idx != -1 and leaving_o_idx != -1: # OH基を削除 rwmol.RemoveAtom(leaving_o_idx) # N末端のNとC末端のCを単結合でつなぐ rwmol.AddBond(n_atom_idx, c_atom_idx, Chem.BondType.SINGLE) # 編集を確定して通常の分子オブジェクトに戻す mol_cyclic = rwmol.GetMol() try: # 化学的な整合性をチェック Chem.SanitizeMol(mol_cyclic) # 水素原子を付加(3D構造生成に必要) mol_h = Chem.AddHs(mol_cyclic) # 4. 3Dコンフォマー(立体構造)の生成 # ETKDGというアルゴリズムを使って3D座標を計算します AllChem.EmbedMolecule(mol_h, AllChem.ETKDG()) return mol_h except Exception as e: print(f"Error generating 3D for {seq}: {e}") return None return None
# ----------------------------------------------------
# STEP 1: リードペプチドの定義
# ----------------------------------------------------
# ここでは探索の起点となるペプチド配列(リードペプチド)を定義します。
# FASTA形式('HMTEVVRRC')からツールで扱いやすいHELM形式に変換しています。
lead_peptide = convert_FASTA_to_HELM('HMTEVVRRC')[0]
print(f"リードペプチド(HELM形式): {lead_peptide}")
# ----------------------------------------------------
# STEP 2: シードライブラリの生成
# ----------------------------------------------------
# リードペプチドを元に、少しだけ配列を変えたペプチド群(ライブラリ)を作ります。
seed_library = [lead_peptide]
# アラニンスキャニング: 各アミノ酸をアラニンに変えたものを作成
for seq in alanine_scanning(lead_peptide): seed_library.append(seq)
# ホモログスキャニング: 各アミノ酸を似た性質のアミノ酸に変えたものを作成
for seq in homolog_scanning(lead_peptide): seed_library.append(seq) # デモ用に数を制限しています(最大10個追加でストップ) if len(seed_library) >= 10: print('Reach max. number of peptides allowed.') break
print(f"Total peptides generated: {len(seed_library)}")
print("First 5 peptides:")
for p in seed_library[:5]: print(p)
# ----------------------------------------------------
# STEP 3: 3D Visualization (3D可視化)
# ----------------------------------------------------
print("Generating 3D structures for the first 3 peptides...")
# 生成したライブラリの最初の3つだけを可視化してみます
for i, helm in enumerate(seed_library[:3]): print(f"\nPeptide {i+1}: {helm}") # 定義した関数を使ってHELM文字列から3D構造データを生成 mol = helm_to_3d_mol(helm) if mol: # py3Dmolを使ってブラウザ上で分子を表示 view = py3Dmol.view(width=400, height=300) # 分子データを渡す view.addModel(Chem.MolToMolBlock(mol), 'mol') # 表示スタイルをスティック(棒)モデルに設定 view.setStyle({'stick': {}}) # 分子が画面に収まるようにズーム view.zoomTo() view.show() else: print("Failed to create 3D structure.")
# ----------------------------------------------------
# STEP 4: PDBQTファイルの保存
# ----------------------------------------------------
from google.colab import drive
import os
# Google Driveをマウント(接続)します。
# これにより、作成したデータを自分のDriveに保存できるようになります。
drive.mount('/content/drive')
# 出力先のディレクトリ(フォルダ)パスを定義します
output_dir = '/content/drive/MyDrive/Cyclic_peptide_screening'
# ディレクトリが存在しない場合は作成します(exist_ok=Trueで既存の場合のエラーを回避)
os.makedirs(output_dir, exist_ok=True)
print(f"Output directory created/verified at: {output_dir}")
from meeko import MoleculePreparation
from rdkit import Chem
import requests
def save_mol_to_pdbqt(mol, name, output_dir): """ RDKitの分子オブジェクトをPDBQT形式で保存する関数です。 PDBQTはドッキングシミュレーション(AutoDockなど)で使われる形式です。 """ try: # Meekoを使って分子の前処理(プレパレーション)を行います # これにより、結合の回転などを設定したPDBQTデータが作られます preparator = MoleculePreparation() preparator.prepare(mol) pdbqt_string = preparator.write_pdbqt_string() # ファイルとして保存 filename = os.path.join(output_dir, f"{name}.pdbqt") with open(filename, 'w') as f: f.write(pdbqt_string) return True except Exception as e: print(f"Error converting {name}: {e}") return False
print("Starting PDBQT conversion for peptide library...")
success_count = 0
# ライブラリ内の全ペプチドについて処理を実行します
for i, helm in enumerate(seed_library): # ファイル名用に配列を取得 seq_name = parse_helm_to_seq(helm) base_name = seq_name if seq_name else f"peptide_{i+1:03d}" name = f"Cyclic_{base_name}" # 3D構造を生成 mol = helm_to_3d_mol(helm) if mol: # 分子に名前プロパティを追加 mol.SetProp("_Name", name) # PDBQT形式で保存 if save_mol_to_pdbqt(mol, name, output_dir): success_count += 1 else: print(f"Failed to save {name}") else: print(f"Failed to generate 3D for {name} ({helm})")
print(f"\nSuccessfully saved {success_count}/{len(seed_library)} peptides to {output_dir}") コード詳細解説
!pip install rdkit py3Dmol
!pip install meeko
!pip install gemmi
import rdkit
from rdkit import Chem
from rdkit.Chem import AllChem
import py3Dmol
from typing import List
import reまずは、以下のように必要なライブラリをインストールします。
!pip install rdkit py3Dmolrdkit:分子構造の読み込み、編集、描画、化学構造に基づく演算(化学フラグメント検索、構造変換など)を行う強力なライブラリ。py3Dmol:3D分子構造をWebベースで可視化するためのインターフェース。3Dmol.jsというJavaScriptライブラリをPythonから操作できるようにしたもの。
💡 rdkit は in silico創薬やQSAR解析で頻繁に使われる定番ライブラリです。
!pip install meekomeeko:AutoDock Vinaの前処理ツールであるprepare_ligandを Python で扱うためのラッパー。- リガンドのフォーマット変換やプロトン化状態の調整、トーションの指定など、分子ドッキング前の処理が行えます。
!pip install gemmigemmi:結晶構造(PDB / mmCIF)ファイルを高速かつ正確に読み書きできるバイオインフォマティクスライブラリ。pdb構造の取り扱いや電子密度マップとの連携にも強く、研究現場での使い勝手が高いです。
モジュールのインポート
次に、インストールしたライブラリをインポートしていきます。
import rdkit
from rdkitimport Chem
from rdkit.Chemimport AllChemrdkit: ライブラリ全体のベース。Chem: 分子(Molオブジェクト)の読み書き、構造の基本操作に使います。AllChem: 3D座標の付加(構造最適化や立体構造生成)や部分構造検索、化学反応の定義など、より高度な機能を提供。
import py3Dmolpy3Dmol: 3D構造をWeb上でインタラクティブに可視化するためのライブラリ。
from typingimportListtypingモジュールのListは、関数やクラスで**リストの型注釈(型ヒント)**を与えるために使います。defprocess_molecules(mols: List[str]):
import rereは **正規表現(regex)**モジュールで、文字列の検索・抽出・置換などができます。- 例えば、分子名からIDや化合物情報を抽出する場面で活躍します。
関数の定義
# --- Helper Functions (補助関数) ---
def convert_FASTA_to_HELM(seq: str, cyclic: bool = True) -> List[str]: """ FASTA形式の配列(例: 'ACDEF')をHELM形式(階層的編集言語)に変換する関数です。 cyclic=True の場合、先頭と末尾を結合して環状ペプチドにします(バックボーン環化)。 """ # 1. 配列の文字をドット区切りにします(例: H.M.T...) polymer = ".".join(list(seq)) # PEPTIDE1という名前のポリマーとして定義します helm = f"PEPTIDE1{{{polymer}}}" if cyclic: # 2. 環状化の処理 # N末端(R1)とC末端(R2)を結合させる記述を追加します n = len(seq) # 接続の構文: SourcePoly,TargetPoly,SourceAtom:Att-TargetAtom:Att connection = f"PEPTIDE1,PEPTIDE1,1:R1-{n}:R2" helm += f"${connection}$$$$" else: # 環状化しない場合は終了記号のみ追加 helm += "$$$$" return [helm]
def parse_helm_to_seq(helm: str) -> str: """HELM文字列からアミノ酸配列のみを抽出する関数です。""" # 正規表現を使って PEPTIDE1{...} の中身を取り出します match = re.search(r'PEPTIDE1\{(.*?)\}', helm) if match: # ドットを取り除いて文字列として返します(例: H.M.T -> HMT) return match.group(1).replace('.', '') return ""
def alanine_scanning(helm_str: str) -> List[str]: """ アラニンスキャニングを行う関数です。 ペプチドの各アミノ酸を1つずつアラニン(A)に置換した変異体を作成します。 特定のアミノ酸が活性に重要かどうかを調べるためによく使われます。 """ seq = parse_helm_to_seq(helm_str) variants = [] for i in range(len(seq)): # 元々がアラニンの場合はスキップ if seq[i] == 'A': continue # i番目のアミノ酸を'A'に置き換えた新しい配列を作成 new_seq = seq[:i] + 'A' + seq[i+1:] # HELM形式に変換してリストに追加 variants.append(convert_FASTA_to_HELM(new_seq)[0]) return variants
def homolog_scanning(helm_str: str) -> List[str]: """ ホモログスキャニングを行う関数です。 各アミノ酸を、性質が似ている別のアミノ酸(ホモログ)に置換した変異体を作成します。 """ seq = parse_helm_to_seq(helm_str) # 置換ルール(辞書): キーのアミノ酸を、値のリストにあるアミノ酸に置換します homologs = { 'H': ['K', 'R'], 'K': ['R', 'H'], 'R': ['K', 'H'], # 塩基性アミノ酸間での置換 'D': ['E'], 'E': ['D'], # 酸性アミノ酸間 'N': ['Q'], 'Q': ['N'], # アミド基を持つもの同士 'S': ['T'], 'T': ['S'], # 水酸基を持つもの同士 'V': ['I', 'L'], 'L': ['I', 'V'], 'I': ['L', 'V'], # 疎水性アミノ酸間 'F': ['Y'], 'Y': ['F'], # 芳香族アミノ酸間 'A': ['G'], 'G': ['A'], # 小さいアミノ酸間 'M': ['L'] } variants = [] for i in range(len(seq)): aa = seq[i] # そのアミノ酸に対する置換ルールが存在する場合 if aa in homologs: for sub in homologs[aa]: # 置換して新しい配列を作成 new_seq = seq[:i] + sub + seq[i+1:] variants.append(convert_FASTA_to_HELM(new_seq)[0]) return variants
def helm_to_3d_mol(helm_str: str): """ HELM文字列をRDKitの分子オブジェクト(3D構造込み)に変換する関数です。 RDKitを使って直鎖ペプチドを作成し、化学的に結合させて環状にします。 """ # 1. 配列情報を取得 seq = parse_helm_to_seq(helm_str) if not seq: return None # 2. 配列から直鎖状のペプチド分子を作成 mol = Chem.MolFromSequence(seq) if mol is None: return None # 3. 環状化(Head-to-Tail cyclization)の処理 # 分子を編集可能な状態(RWMol)にします rwmol = Chem.RWMol(mol) # N末端(アミノ基)とC末端(カルボキシ基)の原子を探します # SMARTS記法というパターンマッチングを使っています n_matches = rwmol.GetSubstructMatches(Chem.MolFromSmarts("[NH2,NH3]")) # N末端 c_matches = rwmol.GetSubstructMatches(Chem.MolFromSmarts("C(=O)[OH]")) # C末端 if not n_matches or not c_matches: print(f"Could not identify termini for {seq}") return None # N末端の窒素原子のインデックス n_atom_idx = n_matches[0][0] # C末端の炭素原子と、脱離するOH基の酸素原子を特定します c_term_indices = c_matches[-1] # 最後に見つかったものをC末端と仮定 c_atom_idx = -1 leaving_o_idx = -1 for idx in c_term_indices: atom = rwmol.GetAtomWithIdx(idx) if atom.GetSymbol() == 'C': # 炭素に結合している酸素(OH)を探す for bond in atom.GetBonds(): neighbor = bond.GetOtherAtom(atom) if neighbor.GetSymbol() == 'O' and bond.GetBondType() == Chem.BondType.SINGLE: c_atom_idx = idx leaving_o_idx = neighbor.GetIdx() break if c_atom_idx != -1: break # ペプチド結合の形成(脱水縮合のシミュレーション) if c_atom_idx != -1 and leaving_o_idx != -1: # OH基を削除 rwmol.RemoveAtom(leaving_o_idx) # N末端のNとC末端のCを単結合でつなぐ rwmol.AddBond(n_atom_idx, c_atom_idx, Chem.BondType.SINGLE) # 編集を確定して通常の分子オブジェクトに戻す mol_cyclic = rwmol.GetMol() try: # 化学的な整合性をチェック Chem.SanitizeMol(mol_cyclic) # 水素原子を付加(3D構造生成に必要) mol_h = Chem.AddHs(mol_cyclic) # 4. 3Dコンフォマー(立体構造)の生成 # ETKDGというアルゴリズムを使って3D座標を計算します AllChem.EmbedMolecule(mol_h, AllChem.ETKDG()) return mol_h except Exception as e: print(f"Error generating 3D for {seq}: {e}") return None return Noneぺプチドライブラリのスクリーニングや構造最適化でよく使われる補助関数群を紹介します。特に以下のような作業を自動化できます:
- ✅ FASTA形式のアミノ酸配列 → HELM形式への変換
- ✅ アラニンスキャニング・ホモログスキャニングの自動化
- ✅ HELM構造からの3D分子生成(RDKit使用)
in silico 創薬やペプチド設計の現場で、高速にスクリーニング候補を生成・検討するための便利ツール群です。
1. convert_FASTA_to_HELM: FASTA配列 → HELM形式へ変換
def convert_FASTA_to_HELM(seq: str, cyclic:bool =True) ->List[str]:- FASTA形式(例:
"ACDEFGHIK")を、HELM形式(例:PEPTIDE1{A.C.D.E.F.G.H.I.K}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$)に変換する関数です。 cyclic=Trueにすると、**環状ペプチド(ヘッド・トゥ・テール環化)**になります。
2. parse_helm_to_seq: HELM文字列 → アミノ酸配列抽出
def parse_helm_to_seq(helm: str) ->str:- HELM形式の中身から、アミノ酸配列部分(例:
"ACD")だけを取り出す関数です。 - 正規表現で
PEPTIDE1{...}の中身を抽出 →"A.C.D"→"ACD"に変換します。
3. alanine_scanning: アラニンスキャニング
def alanine_scanning(helm_str: str) ->List[str]:- 各アミノ酸を アラニン(A)に1つずつ置換 した HELM 配列のリストを生成します。
- アラニンスキャニングは、活性に重要な残基の特定に有効です(SAR解析など)。
例:
元:HELM(ACD) →[A→A, C→A, D→A]
結果:[AAD, AAD, ACA]4. homolog_scanning: ホモログスキャニング
def homolog_scanning(helm_str: str) ->List[str]:- 各アミノ酸を**性質の似たアミノ酸(ホモログ)**に置き換えたバリエーションを生成します。
- 例えば、
K(リジン)→R(アルギニン)に置換、といった操作です。
置換辞書は生理化学的性質に基づいています:
'K': ['R','H'],# 塩基性アミノ酸間
'F': ['Y'],# 芳香族アミノ酸間
...5. helm_to_3d_mol: HELM文字列 → RDKitで3D構造生成
defhelm_to_3d_mol(helm_str: str):この関数は、次の処理をステップバイステップで行います:
▶️ 処理の流れ
- HELM → アミノ酸配列抽出
- 配列から直鎖ペプチドを生成(
Chem.MolFromSequence) - N末端とC末端の原子を検出
- 脱水縮合(OH削除+N-C結合)により環化
- Sanitize(分子構造チェック)+水素追加
- 3D構造生成(ETKDGアルゴリズム使用)
✅ 出力
- 成功すれば
Molオブジェクト(3D座標付き) - 可視化には
py3Dmolやrdkit.Chem.Draw.MolToImageが使えます。
STEP 1: リードペプチドの定義
# ----------------------------------------------------
# STEP 1: リードペプチドの定義
# ----------------------------------------------------
# ここでは探索の起点となるペプチド配列(リードペプチド)を定義します。
# FASTA形式('HMTEVVRRC')からツールで扱いやすいHELM形式に変換しています。
lead_peptide = convert_FASTA_to_HELM('HMTEVVRRC')[0]
print(f"リードペプチド(HELM形式): {lead_peptide}")このステップでやっていること
- 入力:リードペプチドの配列
'HMTEVVRRC'(FASTA形式) - 処理:
convert_FASTA_to_HELM関数を使って、HELM形式 に変換 - 出力:環状ペプチドを表す HELM 文字列
なぜHELM形式に変換するのか?
HELM(Hierarchical Editing Language for Macromolecules)は、以下の理由でペプチドや核酸の表現に適した形式です:
- 修飾残基や環状構造を正確に記述できる
- 分子設計ツール(Schrödinger、BioViaなど)と互換性が高い
- 配列ベースと構造ベースを統合した記述が可能
変換の例
FASTA:'HMTEVVRRC'
↓
HELM: PEPTIDE1{H.M.T.E.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$.join(list(seq))により、アミノ酸がドット(.)で区切られる1:R1-9:R2は、1番目と9番目の残基の末端(R1, R2)を結合 → 環状ペプチド化
STEP 2: シードライブラリの生成
# ----------------------------------------------------
# STEP 2: シードライブラリの生成
# ----------------------------------------------------
# リードペプチドを元に、少しだけ配列を変えたペプチド群(ライブラリ)を作ります。
seed_library = [lead_peptide]
# アラニンスキャニング: 各アミノ酸をアラニンに変えたものを作成
for seq in alanine_scanning(lead_peptide): seed_library.append(seq)
# ホモログスキャニング: 各アミノ酸を似た性質のアミノ酸に変えたものを作成
for seq in homolog_scanning(lead_peptide): seed_library.append(seq) # デモ用に数を制限しています(最大10個追加でストップ) if len(seed_library) >= 10: print('Reach max. number of peptides allowed.') break
print(f"Total peptides generated: {len(seed_library)}")
print("First 5 peptides:")
for p in seed_library[:5]: print(p)このステップでは、STEP 1 で定義したリードペプチドを基にして、構造が少しだけ異なるペプチド群(変異体)を自動で生成します。
seed_library = [lead_peptide]seed_libraryというリストを作成し、まずは元のリードペプチド(HELM形式)を追加します。
アラニンスキャニング(alanine_scanning)
for seqin alanine_scanning(lead_peptide): seed_library.append(seq)- 各アミノ酸を1つずつ アラニン(A) に置換して、活性に重要な残基を探索するための変異体を生成します。
- 例えば、
HMTEVVRRCの中でM→Aとなると、HATEVVRRCのような変異が作られます。
🧪 研究応用:
アラニンスキャニングは、機能性アミノ酸(酵素活性部位や結合ポケットなど)を特定するための標準的な手法です。
ホモログスキャニング(homolog_scanning)
for seqin homolog_scanning(lead_peptide): seed_library.append(seq)
iflen(seed_library) >=10:
print('Reach max. number of peptides allowed.')
break- 各アミノ酸を、性質が似ている別のアミノ酸に置換します(例:
H → KやF → Y)。 - 生理化学的性質が近いので、構造や機能を大きく損なわずにバリエーションを広げられます。
- このコードでは、生成する変異体の数を最大10個に制限しています(チュートリアル用の簡易実装)。
🧪 研究応用:
ホモログスキャニングは、構造保存的な最適化や進化的な置換可能性の評価に役立ちます。
結果の確認
print(f"Total peptides generated: {len(seed_library)}")
print("First 5 peptides:")
for pin seed_library[:5]:
print(p)- 作成したペプチドライブラリの個数を出力し、先頭5件の HELM 表現を表示します。
実行例(出力)
Total peptidesgenerated:10
First5peptides:
PEPTIDE1{H.M.T.E.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$
PEPTIDE1{A.M.T.E.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$
PEPTIDE1{H.A.T.E.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$
PEPTIDE1{H.M.A.E.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$
PEPTIDE1{H.M.T.A.V.V.R.R.C}$PEPTIDE1,PEPTIDE1,1:R1-9:R2$$$$STEP 3: 3D Visualization (3D可視化)
# ----------------------------------------------------
# STEP 3: 3D Visualization (3D可視化)
# ----------------------------------------------------
print("Generating 3D structures for the first 3 peptides...")
# 生成したライブラリの最初の3つだけを可視化してみます
for i, helm in enumerate(seed_library[:3]): print(f"\nPeptide {i+1}: {helm}") # 定義した関数を使ってHELM文字列から3D構造データを生成 mol = helm_to_3d_mol(helm) if mol: # py3Dmolを使ってブラウザ上で分子を表示 view = py3Dmol.view(width=400, height=300) # 分子データを渡す view.addModel(Chem.MolToMolBlock(mol), 'mol') # 表示スタイルをスティック(棒)モデルに設定 view.setStyle({'stick': {}}) # 分子が画面に収まるようにズーム view.zoomTo() view.show() else: print("Failed to create 3D structure.")STEP 3:ペプチドの3D構造を可視化する(py3Dmol)
このステップでは、STEP 2で作成したペプチドライブラリのうち、最初の3つのペプチドについて、立体構造(3D構造)を自動生成し、インタラクティブに可視化します。
print("Generating 3D structures for the first 3 peptides...")処理の流れ
for i, helminenumerate(seed_library[:3]):- 生成済みのペプチドライブラリの先頭3つを対象にループ処理します。
3D構造の生成:helm_to_3d_mol
mol = helm_to_3d_mol(helm)この関数では以下の処理が行われています:
- HELM文字列からアミノ酸配列を抽出(例:
H.M.T...→HMTEVVRRC) RDKitで直鎖ペプチドを生成- N末端とC末端の結合を模倣して環状構造化
- ETKDGアルゴリズムを用いた3D座標生成
ETKDGアルゴリズムとは、分子の3次元構造(3Dコンフォメーション)を生成するための、RDKitで実装された手法のひとつです。正式には「Experimental-Torsion Knowledge Distance Geometry」の略で、距離幾何法(Distance Geometry)をベースに、以下の3要素を統合して高精度な構造生成を実現しています:
- 距離幾何法(DG):原子間の距離制約から3D構造を組み立てる数学的手法。
- 実験トーション角データ(ET):X線結晶構造などから得られた実験値を学習し、現実的な回転角(torsion angles)を生成に反映。
- 立体障害の回避(KDG):原子間の衝突を避け、化学的に自然な配置を目指す。
ETKDGは、SMILESやHELMのような2D構造情報しか持たない分子に、高速かつ現実的な3D構造を付与するための事実上の標準手法です。特に創薬や分子ドッキング、MDシミュレーションの初期構造生成によく用いられます。
- 分子の整合性(Sanitize)と水素の付加
✅ 出力:mol は3D構造を持つ RDKit の Mol オブジェクト
3D可視化:py3Dmol
view = py3Dmol.view(width=400, height=300)
view.addModel(Chem.MolToMolBlock(mol),'mol')
view.setStyle({'stick': {}})
view.zoomTo()
view.show()py3Dmolは Webベースの3D分子可視化ライブラリ(3Dmol.jsのPythonラッパー)- 分子の表示スタイル(スティックモデルなど)を簡単に切り替え可能
- Google Colab や Jupyter Notebook 上で インタラクティブに操作可能
🎯 分子の回転・拡大・色変更など、構造理解が格段にしやすくなります
エラーハンドリング
ifnot mol:
print("Failed to create 3D structure.")- 環状構造の結合エラーや構造生成に失敗した場合でも、スクリプトが止まらずにエラーメッセージを表示します。
- 複雑な構造や端の修飾がある場合は、
helm_to_3d_molが失敗することがあります。
実行結果(例)
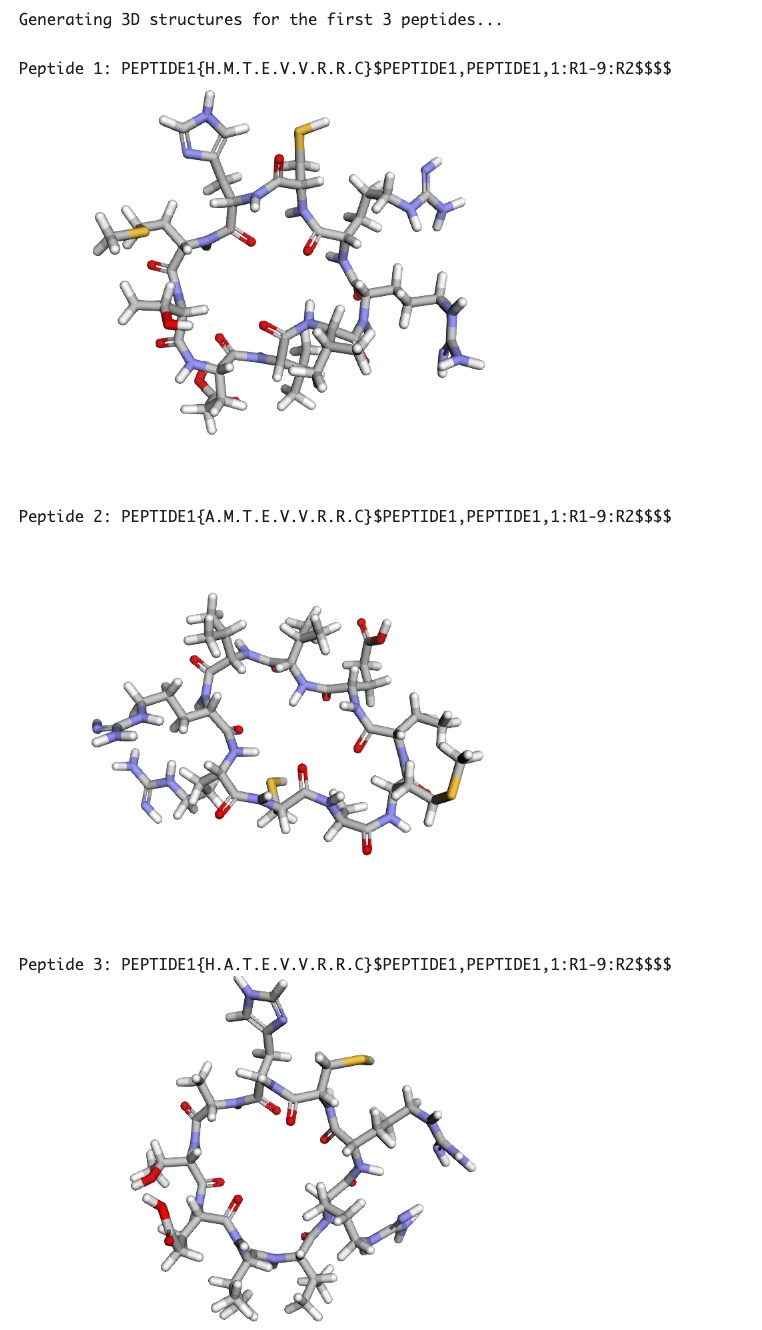
以下のような環状ペプチドが生成されます!
STEP 4: PDBQTファイルの保存
# ----------------------------------------------------
# STEP 4: PDBQTファイルの保存
# ----------------------------------------------------
from google.colab import drive
import os
# Google Driveをマウント(接続)します。
# これにより、作成したデータを自分のDriveに保存できるようになります。
drive.mount('/content/drive')
# 出力先のディレクトリ(フォルダ)パスを定義します
output_dir = '/content/drive/MyDrive/Cyclic_peptide_screening'
# ディレクトリが存在しない場合は作成します(exist_ok=Trueで既存の場合のエラーを回避)
os.makedirs(output_dir, exist_ok=True)
print(f"Output directory created/verified at: {output_dir}")
from meeko import MoleculePreparation
from rdkit import Chem
import requests
def save_mol_to_pdbqt(mol, name, output_dir): """ RDKitの分子オブジェクトをPDBQT形式で保存する関数です。 PDBQTはドッキングシミュレーション(AutoDockなど)で使われる形式です。 """ try: # Meekoを使って分子の前処理(プレパレーション)を行います # これにより、結合の回転などを設定したPDBQTデータが作られます preparator = MoleculePreparation() preparator.prepare(mol) pdbqt_string = preparator.write_pdbqt_string() # ファイルとして保存 filename = os.path.join(output_dir, f"{name}.pdbqt") with open(filename, 'w') as f: f.write(pdbqt_string) return True except Exception as e: print(f"Error converting {name}: {e}") return False
print("Starting PDBQT conversion for peptide library...")
success_count = 0
# ライブラリ内の全ペプチドについて処理を実行します
for i, helm in enumerate(seed_library): # ファイル名用に配列を取得 seq_name = parse_helm_to_seq(helm) base_name = seq_name if seq_name else f"peptide_{i+1:03d}" name = f"Cyclic_{base_name}" # 3D構造を生成 mol = helm_to_3d_mol(helm) if mol: # 分子に名前プロパティを追加 mol.SetProp("_Name", name) # PDBQT形式で保存 if save_mol_to_pdbqt(mol, name, output_dir): success_count += 1 else: print(f"Failed to save {name}") else: print(f"Failed to generate 3D for {name} ({helm})")
print(f"\nSuccessfully saved {success_count}/{len(seed_library)} peptides to {output_dir}") STEP 4:PDBQTファイルの保存(AutoDock用)
このステップでは、STEP 3で生成したペプチドの3D構造を、AutoDock や AutoDock Vina で使える PDBQT形式 に変換し、Google Drive に保存します。
Google Driveとの接続と出力先の設定
from google.colabimport drive
import os
drive.mount('/content/drive')
output_dir ='/content/drive/MyDrive/Cyclic_peptide_screening'
os.makedirs(output_dir, exist_ok=True)drive.mount:Colab と Google Drive を接続します。os.makedirs(..., exist_ok=True):出力フォルダを作成(既にあってもOK)
💡 これで、変換したファイルを 自分のDriveに自動保存できるようになります。
Meeko を使った PDBQT 変換関数の定義
from meekoimport MoleculePreparation
from rdkitimport Chem
def save_mol_to_pdbqt(mol, name, output_dir):この関数は次の処理を自動で行います:
Meekoを使って RDKit 分子をプレパレーション- 回転可能な結合情報などを含む PDBQT文字列を生成
- 指定フォルダに
.pdbqtファイルとして保存
例:
save_mol_to_pdbqt(mol,"Cyclic_HMTEVVRRC","/content/drive/MyDrive/...")ライブラリ全体のバッチ変換処理
for i, helminenumerate(seed_library): mol = helm_to_3d_mol(helm) ... save_mol_to_pdbqt(mol, name, output_dir)- すべてのペプチドに対して
helm_to_3d_mol()を使って 3D構造を生成 - Meeko に渡して
.pdbqt形式で保存
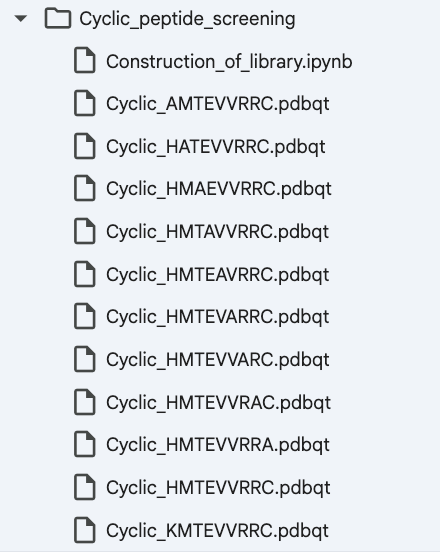
ファイル名は次のようになります:
Cyclic_HMTEVVRRC.pdbqt
Cyclic_AMTEVVRRC.pdbqt
Cyclic_HATEVVRRC.pdbqt
...実行結果のログ出力
print(f"\nSuccessfully saved {success_count}/{len(seed_library)} peptides to{output_dir}")例:
Successfully saved10/10 peptidesto /content/drive/MyDrive/Cyclic_peptide_screening- 保存に成功した数を表示してくれるため、エラー検出や進捗確認が容易です。
結果
上記のコードが全てrunされると、設定したoutputディレクトリに環状ペプチドライブラリが出力されます!
最後に
今日はHELM形式という形式を使って、環状ペプチドライブラリの作成を行いました!
非天然アミノ酸も設定により、構築可能なので、あらゆるライブラリに対応可能です。
ぜひお望みのライブラリを設定してみてください!次は本ライブラリを使って、スクリーニングを実行します!
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

