

本記事は分子ドッキングに使用するAutodock CrankPep(ADCP)について書かれた記事です。今回は環状ペプチドとタンパク質とのドッキングを例にご紹介します
【この記事のまとめ】
環状ペプチド創薬の難関である「高い柔軟性」を考慮したドッキングシミュレーションを実現するため、AutoDock CrankPep(ADCP)を用いた標的タンパク質への結合ポーズ予測手法を詳しく解説します。
- 柔軟なバックボーンのサンプリング: ペプチド主鎖をクランク機構に見立てた独自のアルゴリズム(モンテカルロ法)により、環状構造特有のねじれ角度を効率的に探索し、AutoDock4のスコアリング関数で結合エネルギーを評価。
- WSL/Ubuntuでの環境構築: Windowsユーザー向けにWSL(Windows Subsystem for Linux)の有効化からUbuntu 20.04のインストール、ADCPのセットアップまで、コマンドレベルの具体的な手順を提示。
- 解析結果の定量的評価: 算出された
affinity(エネルギー値)の解釈や、cluster sizeに基づくドッキングポーズの信頼性確認、さらにはPyMOLを用いた視覚的な解析方法を網羅。
この記事を読み進めることで、従来のツールでは困難だった「巨大で柔軟な環状ペプチド」がタンパク質表面のどのポケットに、どのような姿勢で結合するかを、計算機上で高精度に予測・可視化できるようになります。
Windows 11, WSL2(Ubuntu 20.04), Python3.10(Micromamba 管理),bash(Micromamba 初期化済)

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


創薬モダリティとしての環状ペプチドとは?
環状ペプチドは、ペプチド鎖の末端同士を結合して環状構造を形成したモダリティです。比較的に小分子でありながら複雑な立体構造を持ち、直鎖ペプチドと比較して、その立体構造を安定的に保持できることにより、以下の特長を持ちます。
①標的タンパク質との結合において、柔軟すぎない立体構造が有利に働き、高い親和性と選択性を示す。
②安定性が高く、体内の酵素に分解されづらい。
③立体構造の安定性の向上により、膜透過性が上がるとの報告もある。
①の利点により、立体構造を保持しやすいためにタンパク質間相互作用(PPI)を阻害できる点が注目されています。PPI阻害には、比較的広範囲な領域に結合することが求められますが、低分子薬では結合が難しい標的です。環状ペプチドは抗体のように広い表面を認識できるため、これらの「創薬困難標的」に対して新たな選択肢を提供します。
さらに、同じくPPI阻害を行える抗体医薬と比較しても、1~3kDa程度の低分子であることから組織浸透性が高く、培養細胞に頼らずに合成が可能である等の利点を持ち、近年注目が集まっているモダリティです。
Autodock CrankPepとは?
AutoDock CrankPep は、タンパク質–ペプチド複合体の結合モードを予測するためのドッキングツールです。ペプチドの高い柔軟性(主に主鎖のねじれ角度)を考慮しつつ、AutoDock4 のスコアリング関数を用いて相互作用エネルギーを評価します。これにより、ペプチドリガンドがタンパク質表面の溝やポケットにどのようにフィットするかを計算機上で再現できます。
CrankPep アルゴリズムでは、まずペプチドの主鎖を「クランク機構」に見立てた大環状モチーフの回転(モンテカルロ法)で多彩なコンフォメーションを効率的にサンプリングし、側鎖はAutoDockの従来手法で回転自由度を扱います。最終的に複数の探索サイクルを経て得られた構造をクラスタリングし、代表的な結合モデルを出力します。これにより、効率的な予測を可能としており、従来法では時間のかかっていた10~30残基程度の短いペプチドでも、より短時間での予測が可能です。
AutoDock CrankPep は、ペプチドリード化合物の最適化や抗体断片、ミニタンパク質との相互作用解析などに応用され、タンパク質–ペプチド間の創薬標的探索に役立つツールとして広く利用されています。
Autodock CrankPepの環境構築
まずは環境構築をしていきます。
今回はWindowsのWSLを利用します。
- WSL を有効化
- “Windows の機能の有効化または無効化” から「Windows Subsystem for Linux」をオンにして再起動
- Ubuntu(推奨:Ubuntu 20.04)を Microsoft Store からインストールし、起動
- 初回起動時に UNIX ユーザー名とパスワードの設定が求められます
これでwindowsでwslを利用して、linuxコマンドが使えるようになりました。
以上が完了したら、以下のコードで環境構築を行っていきます。
# micromamba をインストールするディレクトリを作成
mkdir -p ~/micromamba_env
# micromamba をインストール(-b: バッチモード, -p: インストール先指定)
curl -Ls https://micro.mamba.pm/install.sh | bash -s -- -b -p ~/micromamba_env
# bash 設定ファイルを読み込み直し(micromamba のパス設定などを反映)
source ~/.bashrc
# ADCP Suite 用の micromamba セットアップスクリプトをダウンロード
wget https://ccsb.scripps.edu/mamba/scripts/adcpsuite_micromamba.sh
# micromamba.sh の読み込みを eval に置き換え(シェルフックを有効にするため)
sed -i 's|\. ${MAMBA_ROOT_PREFIX}/etc/profile.d/micromamba.sh|eval "$(${MAMBA_EXE} shell hook --shell=bash --root-prefix=${MAMBA_ROOT_PREFIX})"|' ./adcpsuite_micromamba.sh
# ADCP Suite の micromamba 環境を構築(依存関係や環境構成を含む)
bash ./adcpsuite_micromamba.sh
# 作成された環境 'adcpsuite' を micromamba でアクティベート
micromamba activate adcpsuiteコードの詳細説明
mkdir -p ~/micromamba_env
mkdir は「フォルダ(ディレクトリ)を作るコマンド」です。
pは「親フォルダもなければ作る」オプション。
~/micromamba_env は「自分のホームフォルダの中にmicromamba_envという名前のフォルダ」を意味します。
ここでやっていること:micromambaを入れるための作業用フォルダを作っています。
例えば、micromambaのプログラムや環境ファイルはここに入ります。
curl -Ls [https://micro.mamba.pm/install.sh](https://micro.mamba.pm/install.sh) | bash -s -- -b -p ~/micromamba_envcurl は「インターネット上のファイルを取得するコマンド」です。
L は「URLがリダイレクトしても追いかける」、-s は「静かに(進捗やエラーを表示しない)」の意味。
ここでは、https://micro.mamba.pm/install.sh というインストール用のスクリプトをネットからダウンロードしています。
|(パイプ)を使って、そのままダウンロードした内容を bash で実行しています。
bash -s -- -b -p ~/micromamba_env の意味は、bashにスクリプトの引数を渡していて、
b は「バイナリモードで(すぐ使える状態で)インストール」
p ~/micromamba_env は「インストール先をさっき作ったフォルダに指定」
ここでやっていること:micromamba本体を指定のフォルダにインストールしています。
source ~/.bashrc~/.bashrc はターミナル(bash)が起動する時に読み込む設定ファイルです。
source は「その設定ファイルを今すぐ読み込む」コマンド。
なぜやるか:micromambaのインストールによって、環境変数(コマンドのパスなど)を設定ファイルに追加しているので、これを反映させてすぐに使えるようにしています。
wget [https://ccsb.scripps.edu/mamba/scripts/adcpsuite_micromamba.sh](https://ccsb.scripps.edu/mamba/scripts/adcpsuite_micromamba.sh)wget もcurlと似ていて、ネットからファイルをダウンロードするコマンドです。
指定のURLからadcpsuite_micromamba.shというスクリプトを取ってきます。
このコードでADCPをダウンロードしています。
sed -i 's|\. ${MAMBA_ROOT_PREFIX}/etc/profile.d/micromamba.sh|eval "$(${MAMBA_EXE} shell hook --shell=bash --root-prefix=${MAMBA_ROOT_PREFIX})"|' ./adcpsuite_micromamba.shsed はテキストファイルの中身を書き換えるコマンドです。
i は「ファイルを直接編集する」という意味。
ここでは、adcpsuite_micromamba.shの中にあるmicromamba初期化コードの書き方を、
古い形式から新しい形式(shell hook方式)に置き換えています。
micromambaの初期化の方法がADCPの作成時から変わっていたので、スクリプトもそれに合わせて直しています。
bash ./adcpsuite_micromamba.shダウンロードして書き換えたスクリプトを実行しています。
このスクリプトの中では、micromambaを使ってADCPの環境構築、必要なパッケージのインストールが行われます。
ADCPをインストール・セットアップしている大事なステップです。
micromamba activate adcpsuitemicromambaで作られた「adcpsuite」という名前の仮想環境(パッケージや設定がまとまった作業用スペース)を「有効化(activate)」します。
これを実行すると、その環境の中に入って、その環境にインストールされたツールやライブラリが使える状態になります。
Autodock CrankPepの実践
以下のTutorialにしたがって行います。
まずは、今回のチュートリアルで使用するreceptorとligandの立体構造のデータをダウンロードしてください。上のリンクの青字の箇所からダウンロードできます(以下の画像の赤で示す箇所です)。
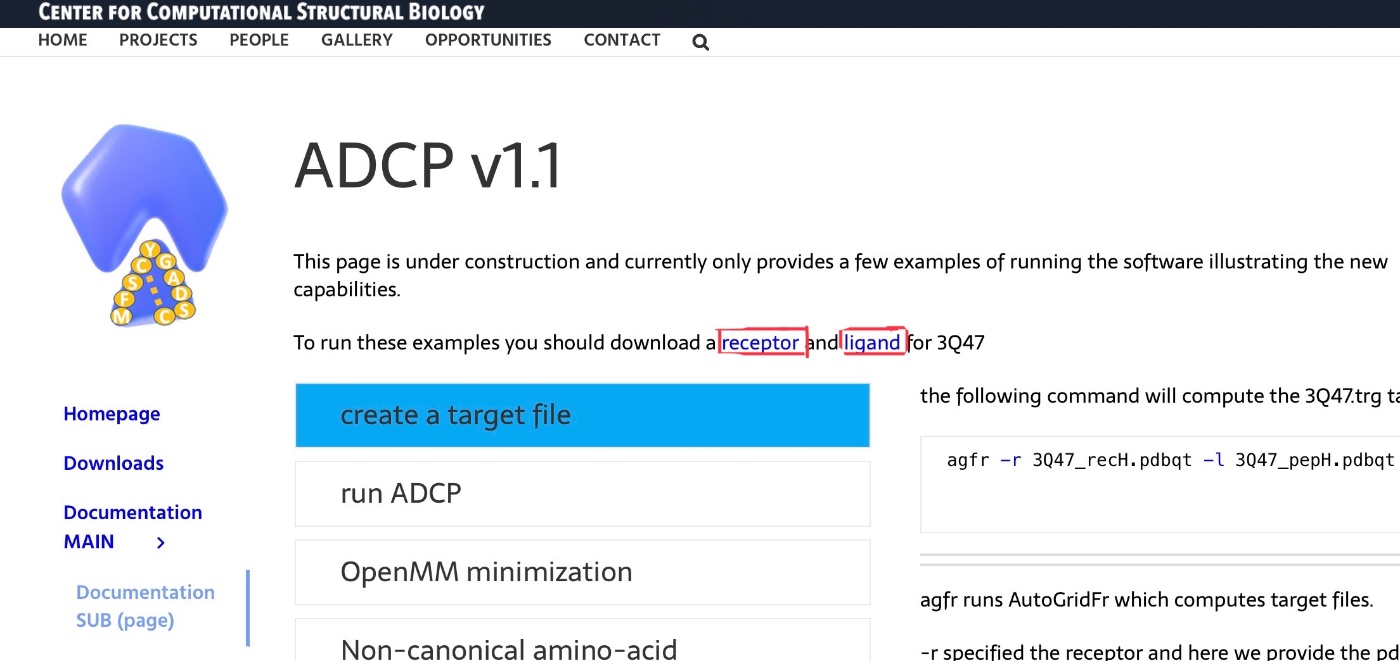
今回は、このレセプターのリガンドが結合する箇所にドッキングするペプチド構造を探していきます。
次に、ダウンロードしたファイルを作業したいフォルダに移動させてください(私はdocuments下に作ったADCPというフォルダに入れています)。
移動させた後は、以下のコマンドで作業したいフォルダに移動します。/documents/ADCPは、ご自身が作業したいフォルダの名前に合わせて変更して下さい。
cd /mnt/c/users/(あなたのWondowsのユーザー名)/documents/ADCPいよいよADCPを使っていきます。
#receptorとligandの立体構造から受容体側の結合箇所を探す
agfr -r 3Q47_recH.pdbqt -l 3Q47_pepH.pdbqt -asv 1.1 -o 3Q47
#直鎖ペプチドとの結合を推定する
adcp -T 3Q47.trg -s "n<BHD>pis&dvd" -N 2 -n 2000 -L swiss -w testDir -o testNSAAandD
#環状ペプチドとの結合を推定する
adcp -O -T 3Q47.trg -s "grctksicfpd" -cyc -N 5 -n 100000 -w testDir1 -o example1 -nmin 5
#optionを追加し、より正確な推定や、溶媒の影響を考慮した推定をする
adcp -O -T 3Q47.trg -s "grctksicfpd" -cyc -N 5 -n 100000 -w testDir1 -o example1 -nmin 5 -nitr 500 -env implicit -dr -reintコードの詳細説明
agfr -r 3Q47_recH.pdbqt -l 3Q47_pepH.pdbqt -asv 1.1 -o 3Q47 は、
受容体(タンパク質)からドッキングできそうな窪みを探し、その周囲に「ドッキング用のボックス」を自動で設定して、ターゲットファイル(.trg)を作るためのコマンドです。実行すると、受容体とボックスの情報をまとめたファイル3Q47.trgが作成されます。
-r 3Q47_recH.pdbqt受容体(タンパク質)ファイル-l 3Q47_pepH.pdbqtリガンド(ペプチド)ファイル-asv 1.1AutoSiteバージョン指定(1.1でOK)-o 3Q47出力名(3Q47.trgファイルが作られる)
adcp -T 3Q47.trg -s "n<BHD>pis&dvd" -N 2 -n 2000 -L swiss -w testDir -o testNSAAandD は、
指定したペプチド(直鎖ペプチド)と受容体とのドッキングをシミュレーションするコマンドです。実行すると、testDirというフォルダが作成され、その中にtestNSAAandD_out.pdb というドッキング結果を含むマルチモデルPDBファイルと、testNSAAandD_summary.dlgというサマリーファイルが作成されます。-T 3Q47.trgターゲット情報(先程で作ったやつ)-s "n<BHD>pis&dvd"ドッキングするペプチドの配列をアルファベット表記したもの。n<BHD>は「アスパラギン(N)」だが、「BHDという修飾基」が側鎖にある。pisは、それぞれプロリン、イソロイシン、セリン。&dはD-アスパラギン酸(D)(&はD体という意味)。 vdは、それぞれバリン、L-アスパラギン酸を表す-N 22回のモンテカルロ探索(今回は計算高速化のため2回で試します)-n 2000それぞれ2000ステップ-L swissスイスロタマーライブラリというデータベースからペプチド側鎖の立体構造を取得-w testDir作業フォルダ-o testNSAAandD出力名(結果ファイル名)
adcp -O -T 3Q47.trg -s "grctksicfpd" -cyc -N 5 -n 100000 -w testDir1 -o example1 -nmin 5 は、
環状ペプチドをドッキングさせ、OpenMMを用いて構造最小化と再評価を行うコマンドです。実行すると、testDir1というフォルダが作られ、その中にexample1_out.pdbという最適な環状ペプチド構造と、そのサマリーexample1_summary.dlgが作成されます。さらに、最適な環状 ペプチドと受容体タンパクの構造がexample1_omm_rescored_out.pdbという名前で作成され、再ランク付けされた各ドッキングソリューションのアンバートポロジと座標ファイルが、testDir1下に作られたexample1_omm_amber_parmというディレクトリ内に生成されます。これらのファイルは、ドッキングポーズのさらなる分析や、分子動力学 (またはその他の分子力学) シミュレーションを使用した自由エネルギー計算に使用できます。
-OOpenMMでのエネルギー評価と最小化を行う-T 3Q47.trgターゲットファイル-s "grctksicfpd"ドッキングするペプチド配列-cyc環状ペプチドであることを指定(N末端とC末端が連結)-N 55回のモンテカルロシミュレーション-n 100000各試行あたり10万ステップ-w testDir1作業ディレクトリ-o example1出力ファイル名の接頭辞-nmin 5上位5構造に対してOpenMM最小化を行う
adcp -O -T 3Q47.trg -s "grctksicfpd" -cyc -N 5 -n 100000 -w testDir1 -o example1 -nmin 5 -nitr 500 -env implicit -dr -reintこれは、さきほどのコマンドに加えて、さらに詳細で現実的な物理的条件を考慮して計算を行うコマンドです。
➕ 追加されたオプションの説明:-nitr 500OpenMMによる最小化(構造調整)を500ステップかけて丁寧に行う(より物理的に正しい構造へ)-env implicit溶媒(主に水)の影響を疑似的に考慮する環境設定。ペプチドが水中にいる状況に近づける。-drドッキング後の構造群(クラスター)に対して、代表構造をさらに再整理する(ドッキングの質を高める)-reintOpenMMを使って**再スコアリング(再評価)**を行う。物理化学的に正しいかどうかを確認。
実行結果
adcp -O -T 3Q47.trg -s "grctksicfpd" -cyc -N 5 -n 100000 -w testDir1 -o example1 -nmin 5上記の環状ペプチドシミュレーションのコードの結果は以下のようになりました。
- example1_summary.dlgの中身
performing MC searches with: /home/yasu/micromamba/envs/adcpsuite/lib/python3.7/site-packages/ADCP/CrankiteAD_Linux-x86_64_1.1
target data from file: 3Q47.trg
job name: example1, summary file example1_summary.dlg, docked poses: example1_out.pdb
Detected 8 cores, using 8 cores.
Performing 5 MC searches using 100000 evals each using a random seed.
Performing search (5 ADCP runs with 100000 steps each) ...
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
Docking performed in 20.85 seconds, i.e. 0 hours 00 minutes 20.850610 seconds
bestEnergies [-3.05982, -2.73379, -7.4937, -4.58936, -2.06522]
bestEnergy in run 3 -7.493700 (0)
Analyzing results ....
concatenating trajectories for run with best energy < 12.506300
concatenated 5 trajectories
Clustering MC trajectories based in contacts using cutoff: 0.800000
finished calculating neighbors for 144 poses with 0.8 seconds
mode | affinity | ref. | clust. | rmsd | energy | best |
| (kcal/mol) | fnc | size | stdv | stdv | run |
-----+------------+------+--------+------+--------+------+
1 -4.4 0.0 7 NA NA 089
2 -4.4 0.0 1 NA NA 084
3 -3.4 0.0 2 NA NA 075
4 -2.7 0.0 6 NA NA 125
5 -2.6 0.0 18 NA NA 071
6 -2.4 0.0 3 NA NA 110
7 -1.8 0.0 4 NA NA 019
8 -1.8 0.0 18 NA NA 109
9 -1.6 0.0 7 NA NA 042
10 -1.4 0.0 7 NA NA 017
11 -1.2 0.0 7 NA NA 132
12 -1.1 0.0 1 NA NA 023
13 -1.0 0.0 2 NA NA 129
14 -0.8 0.0 5 NA NA 010
15 -0.8 0.0 1 NA NA 120
16 -0.8 0.0 3 NA NA 038
17 -0.6 0.0 1 NA NA 054
18 -0.5 0.0 7 NA NA 028
19 -0.3 0.0 1 NA NA 122
20 -0.3 0.0 4 NA NA 043
21 -0.2 0.0 3 NA NA 047
22 0.3 0.0 2 NA NA 020
23 0.3 0.0 1 NA NA 138
24 0.5 0.0 2 NA NA 137
25 0.7 0.0 1 NA NA 133
26 0.8 0.0 1 NA NA 046
27 0.9 0.0 1 NA NA 045
28 0.9 0.0 1 NA NA 027
29 1.0 0.0 1 NA NA 139
30 1.4 0.0 1 NA NA 021
31 1.6 0.0 1 NA NA 136
32 1.6 0.0 1 NA NA 135
33 1.7 0.0 1 NA NA 080
34 2.2 0.0 1 NA NA 044
35 2.2 0.0 1 NA NA 022
36 3.1 0.0 1 NA NA 081
37 4.6 0.0 1 NA NA 024
38 5.4 0.0 1 NA NA 052
39 5.8 0.0 1 NA NA 050
40 6.3 0.0 1 NA NA 116
Calculations completed 22.00 seconds, i.e. 0 hours 00 minutes 20.850610 seconds
MC search command: cd testDir1/example1; /home/yasu/micromamba/envs/adcpsuite/lib/python3.7/site-packages/ADCP/CrankiteAD_Linux-x86_64_1.1 -t 2 "grctksicfpd" -T /mnt/c/users/yasuf/documents/ADCP/testDir1/example1/3Q47 -r 1x100000 -p Bias=NULL,external=5,constrains,1.0,1.0,external2=4,constrains,1.0,1.0,Opt=1,0.25,0.75,0.0 -s 852120 -o run_1.pdb
seed: []
Minimizing docked poses ....
Rescoring clustered poses using OpenMM ..
OpenMM minimization settings: Environment="in-vacuo"; Max_itr=5
OpenMM minimization settings: Cyclizing by backbone
From total 40 models, minimizing top 5 ...
OMM Energy: Working on #1 of 5 models
OMM Energy: E_Complex = -782.88; E_Receptor = -1213.19; E_Peptide = 445.78
OMM Energy: dE_Interaction = -15.47; dE_Complex-Receptor = 430.31
OMM Energy: Working on #2 of 5 models
OMM Energy: E_Complex = -658.82; E_Receptor = -1265.93; E_Peptide = 647.38
OMM Energy: dE_Interaction = -40.27; dE_Complex-Receptor = 607.11
OMM Energy: Working on #3 of 5 models
OMM Energy: E_Complex = 2066.05; E_Receptor = 998.56; E_Peptide = 956.88
OMM Energy: dE_Interaction = 110.61; dE_Complex-Receptor = 1067.49
OMM Energy: Working on #4 of 5 models
OMM Energy: E_Complex = -1971.54; E_Receptor = -2061.98; E_Peptide = 168.10
OMM Energy: dE_Interaction = -77.65; dE_Complex-Receptor = 90.45
OMM Energy: Working on #5 of 5 models
OMM Energy: E_Complex = 674.50; E_Receptor = -330.31; E_Peptide = 1050.35
OMM Energy: dE_Interaction = -45.54; dE_Complex-Receptor = 1004.81
OMM Ranking:REARRANGING output poses using OpenMM (Ecomplex -Ereceptor) energy
OMM Ranking: +<-------OMMscore-------->+<-----------AutoDock CrankPep Scores-------------->+
OMM Ranking:-------+------+------+------------+------------+------------+------+--------+------+--------+------+
OMM Ranking: Model | Rank | Rank | E_Complex | E_Complex | affinity | ref. | clust. | rmsd | energy | best |
OMM Ranking: # |OpenMM| ADCP |-E_Receptor |-E_rec-E_pep| (kcal/mol) | fnc | size | stdv | stdv | run |
OMM Ranking:-------+------+------+------------+------------+------------+------+--------+------+--------+------+
OMM Ranking: 1 1 4 90.4 -77.7 -2.7 0.0 6 NA NA 125
OMM Ranking: 2 2 1 430.3 -15.5 -4.4 0.0 7 NA NA 089
OMM Ranking: 3 3 2 607.1 -40.3 -4.4 0.0 1 NA NA 084
OMM Ranking: 4 4 5 1004.8 -45.5 -2.6 0.0 18 NA NA 071
OMM Ranking: 5 5 3 1067.5 110.6 -3.4 0.0 2 NA NA 075
OMM Ranking:-------+------+------+------------+------------+------------+------+--------+------+--------+------+中盤の表が結合の強さを示す表になっています。
- affinity は結合の良さ(エネルギー値)を示し、一般的に値が小さい(負の値が大きい)ほど良い結合を意味します。
- mode 1 が最も良い(最安定)ドッキングポーズです。
- cluster size は似たポーズがどれだけ集まっているかの指標で、多いほど信頼度が高いこともあります。
また、example1_omm_rescored_out.pdbの中身についてもMol Viewer(https://molstar.org/viewer/)で見でみます。
以下から、example1_omm_rescored_out.pdbを開いてみてください。
受容体のどこに今回の環状ペプチドが結合しているのかが確認できます。

最後に
いかがでしたでしょうか。
実際には、-sでもっと様々な候補ペプチドを指定しながら最適なものを見つけていくのかと思います。ほかのオプションは、計算精度と計算時間のバランスを見ながら試していく感じでしょうか。
以下の論文等も参考に、ぜひ皆さんもトライしてみてください。
https://chemrxiv.org/engage/chemrxiv/article-details/63c7a4da23c13b2e4813aa5b
参考文献
LGPL2 open source
https://www.youtube.com/watch?v=jKHqH_tuLqI
Mol Viewer*
MIT License
GitHub – molstar/molstar: A comprehensive macromolecular library
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

