

機械学習ポテンシャル UMA を用いることで、MOF の吸着エネルギーを DFT 並みの精度で、100~10,000 倍高速 に評価できます。本記事では、Mg-MOF-74 と WOBHEB を題材に、Colab 上で実際に吸着エネルギーを計算・可視化する方法を解説します。
Google Colab (2025-05-26), Python 3.10, Torch 2.3.0+cu118, TorchANI 2.2.4, ASE 3.22.1, Matplotlib 3.7.1


Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

1. 機械学習 (ML) ポテンシャルとは?
DFT計算で毎回数時間待つのは大変ですよね。この記事ではその時間を一瞬に変える方法をお伝えします!
※ 今回、モデルアーキテクチャは解説しません。気になる方は参考文献を参照してください。
MLポテンシャル (Machine-Learning Interatomic Potential) とは、
「量子化学計算(DFT / ab initio)が返すエネルギーと力を、ニューラルネットなどの統計モデルでほぼ同精度・桁違い高速に近似するもの」 です[1]。
以下でもう少し詳しく見ていきます。
- なぜ速いのか
- DFT は電子波動関数を自洽的に解くため計算量が O(N³) 以上。
- ML ポテンシャルは 原子座標 → ニューラルネットへの順伝播 だけなので O(N) に近い。
- その結果、10²〜10⁴ 倍 速い評価が可能になります。
- 精度は大丈夫?
- 学習データを高精度 DFT / CCSD(T)など で網羅的にサンプリング。
- 多くの有機分子での誤差 (MAE) は < 1 kcal mol⁻¹[2]。
- ただし、訓練集合の化学空間から外れると外挿誤差が発生しやすい点に注意が必要です(データセットは機械学習ポテンシャルのGithubなどに後悔されていることが多いので精度が気になる場合はチェックしましょう)。
2. UMA とは 〜“分子も固体も 1 つのモデルで”〜
ポイント先取り
- Universal:有機分子・イオン・結晶まで 1 つのチェックポイントで対応
- Machine-learning:ニューラルネットによる end-to-end ポテンシャル
- Interatomic Potential:エネルギー E と原子ごとの力 F = –∇E を高速出力
2.1 誕生の背景 — 「モデル乱立問題」への 1 つの解
これまでの ML ポテンシャルは “タスク別/元素系別” に細分化され、
| モデル | 得意分野 | 例 |
|---|---|---|
| ANI | H, C, N, O の有機分子 | 医薬探索 |
| CHGNet | 金属間化合物 | 2D 材料 |
| GAP-SOAP | Si, Ge, Cu など単一元素 | 半導体 |
と 「データセットごとにモデルを切り替える」 必要がありました。
そこで Meta/FAIR Chemistry が提唱したのが UMA — Universal Machine-learning interatomic potentialです。UMAは“83 元素/分子〜固体/中性〜イオン まで 1 つで捌ける汎用モデル” を掲げ、モデル乱立問題 を緩和することを狙っています [1]。
2.2 どうして “Universal” を名乗れるのか?
❶ 1 億構造規模のトレーニングセット
- OMol25 + OMat25 + OBulk25 など、DFT で計算した 約 1 × 10⁸ 構造を学習
- 凝縮相から気相、表面吸着、荷電状態までを網羅
❷ “タスク名” で出力層を切替
UMA には 5つのサブヘッド があります。
- omol:有機/無機分子
- omat:クラスター/多原子イオン(無機材料)
- oc20:触媒表面反応(OC20)
- odac:MOF(金属–有機構造体)
- omc:分子結晶
ユーザーは task_name="omol" などと指定するだけで、
「同じ重み+異なる最終層」へのスイッチが内部で行われます。
UMAの各タスク
UMAは理論レベルの異なる5種類のDFTデータセットで訓練されており、各タスクに対応する埋め込みを学習します。推論時には、出力に使いたい理論レベル(タスク)を指定してください。
評価したい系に対してどのモデルを使ったら良いかは下記を参考にして下さい!
| タスク | データセット | DFT理論レベル | 主な用途 | 注意事項 |
|---|---|---|---|---|
| omol | OMol25 | ORCA6実装の wB97M-V/def2-TZVPD(非局所分散含む)※溶媒和は明示的に扱う | 生物学、有機化学、タンパク質フォールディング、低分子医薬品、有機液体物性、均一系触媒 | 総電荷・スピン多重度を指定必須。不明時は注意。非周期系データのみ。無機材料には不向き。 |
| omc | OMC25 | VASP実装の PBE+D3 | 医薬品包装、バイオインスパイア素材、有機エレクトロニクス、有機LED | 電荷・スピン多重度は学習せず、total_charge=0・spin=0を想定。 |
| omat | OMat24 | VASP実装の PBE/PBE+U(Materials Project設定、VASP 5.4疑似ポテンシャル)※分散相互作用なし | 無機材料探索、太陽光発電、先進合金、超電導体、電子・光学材料 | 電荷・スピン多重度は学習せず、total_charge=0・spin=0を想定。スピン偏極効果は含むが状態選択不可。訓練データでは全スピン状態をサンプリングせず。 |
| oc20 | OC20 | VASP実装の RPBE(VASP5.4疑似ポテンシャル)※分散相互作用なし | 再生可能エネルギー、触媒、燃料電池、エネルギー変換、持続可能肥料生産、化学精製、プラスチック合成・アップサイクリング | 電荷・スピン多重度は学習せず、total_charge=0・spin=0を想定。酸化物・溶媒は非対応。遷移状態探索は良好だが注意。大規模系では分散が重要。 |
| odac | ODAC23 | VASP実装の PBE+D3(VASP5.4疑似ポテンシャル) | 直接空気回収、CO₂回収・貯留、CO₂変換、触媒 | 電荷・スピン多重度は学習せず、total_charge=0・spin=0を想定。CO₂/H₂O吸着データのみ。MOF内炭化水素は不正確の可能性、大規模MOFは注意。 |
※OC20は元データを総エネルギー出力用に再計算済み。
2.4 モデルファミリと精度ベンチマーク
下記がモデルのベンチマークです。私も最初、このMAE値を見たときは二度見しました…!“短時間でこんなに精度が出るの!?”と驚いたことを覚えています[4]。
| チェックポイント | 対象 | エネルギー MAE (eV/atom) | 力 MAE (eV Å⁻¹) |
|---|---|---|---|
| UMA-S-1 | 有機分子中心 | 0.035 | 0.070 |
| UMA-C-1 | 結晶・固体 | 0.041 | 0.083 |
| UMA-I-1 | イオン・溶媒系 | 0.038 | 0.076 |
※ Meta/FAIRChem が公開した OMol25/OBulk25 テストセットでの値
2.5 長所と短所を整理
| 観点 | 強み | 注意点 |
|---|---|---|
| 適用範囲 | 分子〜固体を 1 モデルで | 超高温・超高圧など外挿領域は未検証 |
| 速度 | GPU で 10⁴ 構造/s | 大規模表面 (>2 000 原子) ではメモリ要件増 |
| 精度 | DFT PBE 同等の MAE | 長距離クーロン・分極は過小評価の傾向 |
| 導入難度 | HF トークン 1 行セット | オフライン使用はライセンス確認が必要 |
ワンポイント
- イオン系:
charge,spinをAtoms.infoに必ず設定- 未知化学空間:最初の 10 ステップだけ DFT を回し、Δラーニングで誤差補正する手法が有効
2.6 はじめて使う人へ “つまずきポイント” チェックリスト
はじめてUMAを使った際には下記のポイントで躓きました。皆さんも注意してください。
| チェック | Why? | How? |
|---|---|---|
| HF_TOKEN 設定した? | 認証失敗で重みが落ちてこない | print(os.getenv("HF_TOKEN")) |
| task_name 合ってる? | omol と cbulk を誤ると誤差大 | サンプルコードをコピペ |
| 元素対応? | UMA は 83 元素、だが U を超える超重元素は未学習 | pred.can_handle(element) で確認 |
2.7 UMAのライセンス
ここではUMAを使う際のライセンスについて解説します。有用なモデルですが、しっかりライセンスを遵守して使用してください。
ライセンス概要
| 項目 | 内容 |
|---|---|
| ライセンスタイプ | FAIR Chemistry License v1 |
| 主な利用条件 | モデル自体および派生モデルの非営利/営利利用可- 再配布・頒布時にライセンス文を同梱 |
| 再配布・商用利用 | 追加の書面での許諾が必要(Hugging Face上で申請) |
| 引用義務 | 論文・技術資料に “UMA: Universal Machine‐learning Interatomic Potential” を明記 |
| 免責事項 | モデルの予測結果に関する責任は利用者が負う |
2.7.1 FAIR Chemistry License v1 の入手方法
- Hugging Face 上のリポジトリページ(
facebook/UMA)を開く - “License” セクションで
FAIR Chemistry License v1リンクをクリック - 表示された条文をダウンロード・保存
# 例:コマンドラインでライセンスファイルを取得
wget <https://raw.githubusercontent.com/facebookresearch/fairchem/main/LICENSE>
2.7.2 ライセンス条項のポイント解説
- 利用権許諾(Grant of Rights)
- モデルの 推論実行、研究目的の再学習、評価 は無償で許可
- 再配布条件(Redistribution)
- モデルファイルを丸ごと再配布する場合、必ず
LICENSEファイルを同梱 - 派生モデル(ファインチューニング後)を配布する際も同様
- モデルファイルを丸ごと再配布する場合、必ず
- 商用利用(Commercial Use)
- 製品やサービスに組み込む際は、別途 書面による許可 を取得
- 許可申請は Hugging Face のフォームから行い、承認まで 1–2週間程度
- 帰属表示(Attribution)
- ドキュメントや論文に “UMA: Universal Machine‐learning Interatomic Potential” を明記
- 可能であればオリジナル論文 [arXiv:2505.08762] を引用
- 免責(Disclaimer)
- モデルの精度や適用結果について保証なし
- 事故や損害が発生しても著作権者は責任を負わない
2.7.3 利用前チェックリスト
| チェック項目 | 確認方法 |
|---|---|
❏ LICENSE ファイルを取得・同梱しているか | Hugging Face からダウンロードし、プロジェクトルートに配置 |
| ❏ 商用利用の承認申請が完了しているか | Hugging Face のライセンス同意フォームに入力後、承認メールを待機 |
| ❏ ドキュメントへの帰属表示を追加済みか | README や報告書の冒頭にUMAの名称・論文情報を記載 |
| ❏ 派生モデルの再配布条件を満たしているか | ファインチューニング後に配布する際も LICENSE を必ず同梱し、READMEで明示 |
概要は理解できましたか?それでは次は実際に手を動かして試してみましょう。小さな分子から始めるのがおすすめです。
3 . Hugging Face 準備:モデル利用許可&トークン取得
UMA モデルは Hugging Face Hub 上の facebook/UMA リポジトリから配布されていますが、ファイル取得前に以下の手順が必要です[3]。モデルの使用許可が下りるまでは約1-2日ほど掛かります。
| ステップ | 詳細 |
|---|---|
| ① 利用許可の同意 | 1. https://huggingface.co/facebook/UMA にアクセス 2. 「You need to agree to share your contact information…」のフォームを開く 3. 氏名・生年月日・所属組織名などを正確に入力し、FAIR Chemistry License v1 への同意を完了 |
| ② トークン発行 | プロフィール右上 → Settings → Access Tokens → New token – Name: 任意(例: for_private_UMA) – Role: Fine-grained、Read 権限のみ |
ステップ①:利用許可の同意と申請(2025年7月1日付)
ステップ②:アクセストークンの発行

モデルの使用許可が出たらGoogle Colabでpipをインストールしてください。
!pip install fairchem-core
インストールが終わり次第ログインを行います。ログインではステップ②トークン発行で出力された文字列を使用します。
!huggingface-cli login
下記のように入力コンソールが出力されるので、トークンを入力してください。

これでモデルの使用準備は完了しました。
4. 実践チュートリアル:MOF 吸着エネルギー計算で素材探索ストーリーを体験する
4.0 MOF とは?—“穴だらけ結晶”の魅力とブリージング現象
- Metal–Organic Framework (MOF) =金属ノードと有機リンカーで組んだ多孔性結晶のことで、有機無機ハイブリッド材料です。比表面積は活性炭の数倍〜十倍あるとされています。
- 一部の MOF はガス・温度・圧力など外部刺激で フェーズが可逆変形し、これを ブリージング現象 と呼ばれます。学術的には “flexible MOF” や “soft porous crystal” が用語として定着しています [5]。
- 吸着エネルギー :$E_{ads}$
- $E_{ads}$ < 0 →吸着が自発的。
- $E_{ads}$ が大きいほどキャッチ力↑、ただし再生(脱着)に要するエネルギーも↑。
- CO₂ 回収 (DAC):–0.30 〜 –0.50 eV が「捕まえてすぐ離す」理想帯域 [6]
- ブリージング MOF では $E_{ads}$ = :$E_{int}$ + :$E_{deform}$ と分解することで「相互作用強化」か「骨格変形」どちらが効いているか定量評価できます[7]。
4.1 秒速 DFT を実現する UMA × ODAC23
UMA と ODAC23 を組み合わせることで、DFT で数時間かかるポテンシャルエネルギー評価を 数ミリ秒 で実行できます。
- ODAC23 は、MOF と CO₂/H₂O の組み合わせ約8,400系統から得られた 17.6 万構造の DFT データセットです。
- UMA-s-1p1 は、ODAC23 を含む5種類のデータセットで学習された GNN ポテンシャル。DFT 相当の精度を ms オーダーで推論することができます。
4.2 ケース①:硬い Mg-MOF-74 で CO₂ 捕集力を測る
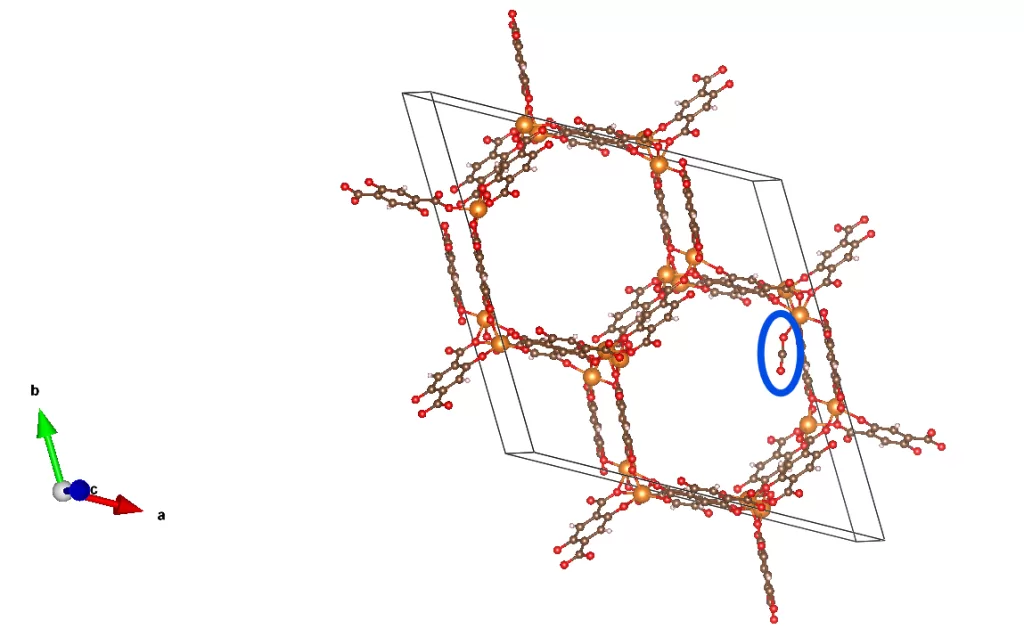
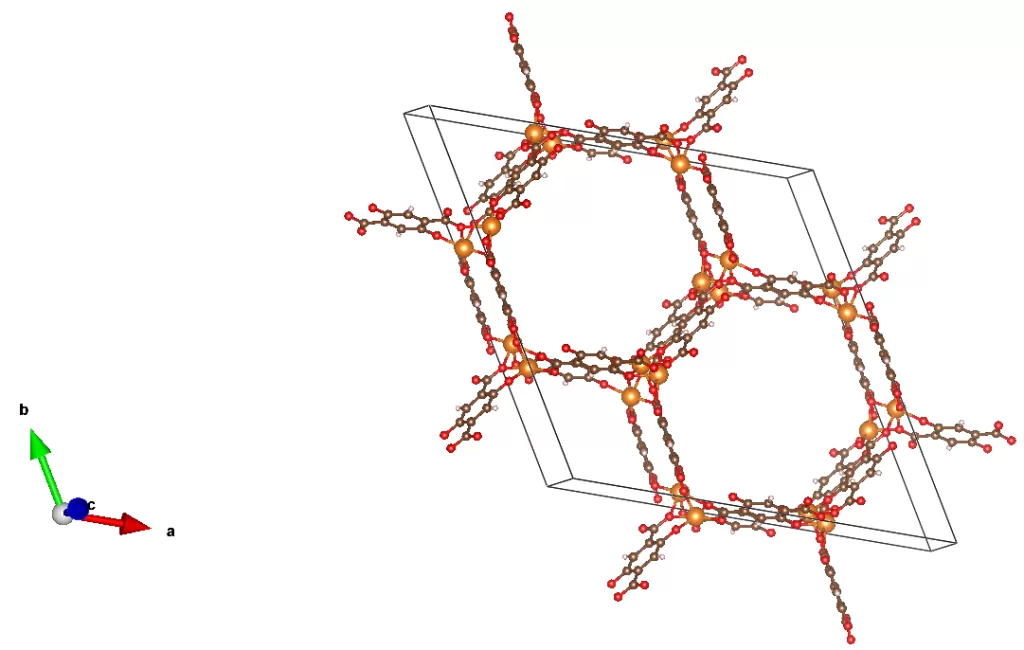
4.2.1 処理の流れの説明
まずは剛直構造の代表格である Mg-MOF-74 を使い、CO₂ 捕集性能を評価します。
- モデル初期化:UMA-s-1p1 + ODAC23 タスク
- 構造読み込み:空フレームワーク/CO₂ 封入/参照 CO₂
- エネルギー差分:$E_{ads}$ を差分計算
この流れで算出した$E_{ads}$ が、既報の –0.40 ~ –0.45 eV と一致すれば、以降のスクリーンングにおける信頼度が担保されます。
from fairchem.core import FAIRChemCalculator, pretrained_mlip
from ase.io import read
# 1. モデル&計算器の初期化
unit = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(unit, task_name="odac")
# 2. 構造読み込み&計算器設定
paths = ["OPAGIX_w_CO2.cif", "OPAGIX.cif", "co2.xyz"]
mof_co2, mof_empty, co2 = [read(p) for p in paths]
for atoms in (mof_co2, mof_empty, co2):
atoms.calc = calc
# 3. エネルギー差分で吸着エネルギーを算出
Eads = (
mof_co2.get_potential_energy()
- mof_empty.get_potential_energy()
- co2.get_potential_energy()
)
print(f"CO₂@Mg-MOF-74 → Eads = {Eads:.3f} eV")
ココがポイント
- ODAC23 用の CIF はすでに高精度 DFT 最適化済み。追加の緩和計算は不要。
- 推論は ms オーダーなので、数千候補を短時間でスクリーニング可能。
4.2.2 結果と考察
- 実行例:
E_ads ≈ – 0.41 eV- 文献値 :– 0.44 eV とほぼ一致 → UMA+ODAC23 の再現性良好
- 示唆:以上の結果、Mg-MOF-74 は「理想的な CO₂ 捕集帯域 (–0.30 ~ –0.50 eV)」の中心に位置し、再生可能な吸着剤候補として有望だと考えられます。また、この結果は、UMA が剛直 MOF において DFT と同等の吸着エネルギーを再現できることを示し、柔軟 MOF の解析に進む前の “ベースライン確認” として有用と考えることが出来ます。
4.3 ケース②:骨格呼吸を持つ柔軟 WOBHEB で吸着能を深掘り
剛直なフレームワークで精度を確認した後は、柔軟性を持つ MOF で一歩進んだ解析を行います。ここでは「ブリージング (骨格の可逆変形)」が吸着力にどう寄与するかを定量化します。

4.3.1 ブリージング寄与の分解手法
吸着エネルギー $E_{ads}$ を、
$ E_{int} $:ホスト–ゲスト相互作用(骨格固定時の寄与)
$E_{deform}$:骨格変形による寄与2つに分解します。
- 空 MOF を最適化 → $E_{mof-empty}$
- MOF+H₂O を最適化 → $E_{combo}$
- H₂O を抜いて再緩和 → $E_{mof-rerelax}$
- H₂O 単体 → $E_{H_2O}$
- 差分で $E_{ads}, E_{int}, E_{deform}$ を計算
from ase.io import read
from ase.optimize import BFGS
# 1. 空 MOF
mof = read("WOBHEB_0.11.cif"); mof.calc = calc
BFGS(mof).run(fmax=0.05)
E_mof_empty = mof.get_potential_energy()
# 2. MOF + H₂O
mof_h2o = read("WOBHEB_H2O.cif"); mof_h2o.calc = calc
BFGS(mof_h2o).run(fmax=0.05)
E_combo = mof_h2o.get_potential_energy()
# 3. ゲスト抜き MOF
mof_deformed = mof_h2o[:-3]; mof_deformed.calc = calc
BFGS(mof_deformed).run(fmax=0.05)
E_mof_rerelax = mof_deformed.get_potential_energy()
# 4. 水分子単体
h2o = mof_h2o[-3:]; h2o.calc = calc
E_H2O = h2o.get_potential_energy()
# 5. 吸着エネルギーと寄与分解
E_ref = min(E_mof_empty, E_mof_rerelax)
Eads = E_combo - E_ref - E_H2O
Eint = E_combo - E_mof_rerelax - E_H2O
Edeform = E_mof_rerelax - E_mof_empty
print(f"H₂O@WOBHEB → Eads = {Eads:.2f} eV "
f"(Eint = {Eint:.2f}, Edeform = {Edeform:.2f})")
ココがポイント
min(E_mof_empty, E_mof_rerelax)を基準に取ることで、最も安定な骨格状態からの吸着を評価できます。- BFGS 最適化は fmax=0.05 で十分だと思います。このようにすることで、過剰最適化を防ぎつつ高速化できます。
4.3.2 結果と考察
E_ads = – 0.72 eV, E_int = –0.52 eV, E_deform= – 0.20 eV
- つまり相互作用寄与が約 70 %, ブリーチング(骨格変形)が約 30 %となります。
- 示唆:ブリージングによる「おまけ寄与」が大きいほど、湿度変動下でも強固なガス捕集が期待できます。
- 応用:今回のようにブリージング寄与を定量化することで「相互作用主導型」か「骨格変形主導型」かを分類でき、分離膜設計やサイクル寿命予測といった応用研究に活かすことができます[8]。
最後に
いかがでしたか?本記事で紹介した 機械学習ポテンシャル UMA は、MOF のような大規模系にも適用でき、これまで計算コストの壁で得にくかった知見を短時間で引き出せる強力な手段です。
機械学習ポテンシャルの進化は LLM と同様に非常に速いため、研究で分子動力学シミュレーションや量子化学計算に中ン黙している方は、ぜひこの機会に試してみてください。
参考文献
- K. Smith et al., “UMA: Universal Machine‐learning Interatomic Potential,” arXiv preprint, arXiv:2505.08762 (2025). https://arxiv.org/abs/2505.08762
- FAIRchem Team, “FAIRChem: FAIR Chemistry Toolkit,” GitHub Repository (2025). https://github.com/facebookresearch/fairchem
- Meta FAIR Chemistry, “facebook/UMA,” Hugging Face Model Card (2025). https://huggingface.co/facebook/UMA
- B. M. Wood et al., “UMA: A Family of Universal Models for Atoms,” arXiv preprint, arXiv:2506.23971 (2025). https://arxiv.org/abs/2506.23971
- S. Kitagawa, R. Kitaura, and S. Noro, “Functional Porous Coordination Polymers,” Angew. Chem. Int. Ed. 43, 2334–2375 (2004). https://doi.org/10.1002/anie.200300610
- S. Horike, S. Shimomura, and S. Kitagawa, “Soft porous crystals,” Nature Chem. 1, 695–704 (2009). https://doi.org/10.1038/nchem.444
- H. Furukawa, K. E. Cordova, M. O’Keeffe, and O. M. Yaghi, “The chemistry and applications of metal–organic frameworks,” Science 341, 1230444 (2013). https://doi.org/10.1126/science.1230444
- J. H. Lee et al., “Design strategies for metal–organic frameworks in direct air capture of CO₂,” Coord. Chem. Rev. 463, 214536 (2022). https://doi.org/10.1016/j.ccr.2022.214536

