

本記事は、NiO(100) 表面上の Pt ナノクラスターに CO/CO₂ を吸着させた触媒モデルを例に、CIF を主軸に機械学習ポテンシャル用の構造を整える最小ワークフローをまとめた実践ガイドです。Google Colab 上で ASE / pymatgen を使ってスラブとクラスターを組み立て、直交化・スーパーセル展開・吸着分子の配置・幾何チェック・可視化を経て「そのまま UMA(Universal Machine-learning interatomic potential)で単点計算に回せる CIF」を出力するまでを解説し、最後に他の表面・クラスター・分子へ応用するための差し替えポイントも整理します。
【この記事のまとめ】
マテリアルズ・インフォマティクス(MI)や計算化学に携わる研究者・学生に向けて、Google Colabを用いた金属触媒の分子モデリングから機械学習ポテンシャル(MLP)計算までの実践的なワークフローを解説します。
- 複雑な触媒モデルの構築:ASEやpymatgenを活用し、NiO表面スラブへのPtナノクラスター担持やCO分子の吸着モデルをコードで正確に作成する手法を紹介。
- 計算精度を高める構造整形:斜格子から直交セルへの変換、スーパーセル展開、衝突チェックなど、シミュレーションの失敗を防ぐためのデータ処理プロセスを詳述。
- MLPによる即時計算:汎用型機械学習ポテンシャル「UMA」を用いて、一点エネルギー計算にそのまま利用可能なCIFファイルを出力・活用するまでの最短ルートを習得。
この記事を読むことで、手作業では困難な触媒構造の作成を自動化し、機械学習を用いた高速・高精度な材料探索の基盤を構築できるようになります。
Google Colab (2025-05-26), Python 3.10, Torch 2.3.0+cu118, TorchANI 2.2.4, ASE 3.22.1, Matplotlib 3.7.1


Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!


0. NiO/Pt ナノクラスター × CO₂還元の準備(セットアップ編)
0-1. なぜ「NiO 上の Pt ナノクラスター × CO₂還元」なのか
CO₂を電気や水素で 燃料・化成品に変える CO₂還元(CO₂RR, CO₂ hydrogenation)は、
「温室効果ガスを減らしつつ、有用な分子を作る」技術として世界中で研究されています。
その中で、多くの理論・実験研究が
- 表面に吸着した CO(*CO)が、CO₂RR の カギ中間体 になっている
- 触媒の性能は、しばしば 「CO の吸着エネルギー」 で整理できる
ことを示しています。
一方、NiO や Ni 系酸化物の上に担持した Pt ナノ粒子は、CO₂の水素化・還元反応で高い活性を示し、
「Pt ナノ粒子 × 酸化物界面」自体が重要な反応サイト になっていることが、
in-situ 分光や理論計算から報告されています。
この記事では、この流れを「CIF 主軸+機械学習ポテンシャル」という観点から整理しなおします。
具体的には、
NiO(100) 表面の上に Pt₁₃ icosahedron ナノクラスター を担持したモデル(NiO/Pt₁₃)を作り、
その上に CO を吸着させて、
「CO₂還元の中での CO 中間体の吸着エネルギー」 を UMA でスキャンする
という位置づけにします。
コード上は CO 分子を扱いますが、
CO₂ → *CO →(C₁/C₂ 生成物)
という CO₂還元の王道メカニズムのなかで、
*「NiO(100)/Pt₁₃ 上で CO がどれくらい安定か」を見る
と考えていただければ OK です。
0-2. このノートで目指すゴール
前回の MOF 編(CIF から UMA 単点まで) と同じく、今回も
「自分で構築した構造を、そのまま UMA で一点エネルギー計算に回せる CIF として仕上げる」
ことをゴールにします。Colabで分子モデリングからMLPで計算【MOF編】
NiO/Pt 編でやることをざっくり書くと:
- NiO rocksalt から NiO(100) 表面スラブ を作る
- 斜格子のスラブを、直交セル+スーパーセル に整える
- その上に Pt₁₃ icosahedron ナノクラスター を“それっぽい距離”で載せる
- 後の章で CO を何パターンか置き、UMA で *CO 吸着エネルギーをスキャンする
0章では、その前提となる 環境構築と共通ユーティリティ だけ先に整えてしまいます。
(実際に NiO や Pt を作るのは 1章以降で行います。)
0-3. 動作環境とライブラリのインストール
今回も Google Colab を想定しています。
使う主なライブラリは:
- ASE:結晶・表面・クラスター作成、CIF の入出力、簡単な可視化
- pymatgen:ASE との相互変換、後で AdsorbateSiteFinder を使う下準備
- fairchem-core:機械学習ポテンシャル UMA を呼び出すためのライブラリ
- huggingface-cli:UMA モデル取得のためのログイン
まず、次のセルを上から順に実行します。
!pip -q install ase pymatgen fairchem-core
!huggingface-cli login- Hugging Face のトークンを聞かれるので、アカウントを持っていない場合は先に作成しておきます。
- 一度ログインしておけば、そのセッションでは UMA をそのまま使えます。
0-4. 共通 import とユーティリティ関数
以降の章で毎回同じコードを書くのは大変なので、
最初に「共通の道具箱」をまとめて定義しておきます。
# 共通import & ユーティリティ
import os, json, csv, glob, hashlib, numpy as np
from pathlib import Path
import matplotlib.pyplot as plt
from ase.io import read, write
from ase.visualize.plot import plot_atoms
from ase.build import bulk, surface, molecule
from ase.cluster.icosahedron import Icosahedron
from ase.geometry import get_distances
from pymatgen.io.ase import AseAtomsAdaptor
from pymatgen.core import Structure
SNAP = Path("snapshots"); SNAP.mkdir(exist_ok=True)
COOR = Path("coords"); COOR.mkdir(exist_ok=True)
def snapshot(atoms, name, rotation='25x,35y,0z', radii=0.9, uc=2, dpi=240, figsize=(5,5)): fig, ax = plt.subplots(figsize=figsize, dpi=dpi) plot_atoms(atoms, ax, rotation=rotation, radii=radii, show_unit_cell=uc) ax.set_axis_off(); plt.tight_layout() out = SNAP / f"{name}.png" plt.savefig(out, bbox_inches="tight", pad_inches=0.02); plt.close(fig) print(f"[PNG] {out}")
def export_coords(atoms, name, fractional=False): cell = atoms.cell.array; pos = atoms.get_positions() if fractional: inv = np.linalg.inv(cell.T); xyz = pos @ inv; header = ["index","symbol","fx","fy","fz"] rows = [(i, atoms[i].symbol, *xyz[i].tolist()) for i in range(len(atoms))] csvp, jsonp = COOR/f"{name}_frac.csv", COOR/f"{name}_frac.json" else: header = ["index","symbol","x","y","z(Å)"] rows = [(i, atoms[i].symbol, *pos[i].tolist()) for i in range(len(atoms))] csvp, jsonp = COOR/f"{name}_cart.csv", COOR/f"{name}_cart.json" with open(csvp,"w",newline="") as f: w=csv.writer(f); w.writerow(header); w.writerows(rows) with open(jsonp,"w") as f: json.dump({"cell":cell.tolist(),"pbc":atoms.get_pbc().tolist(),"header":header,"coords":rows}, f, indent=2) print(f"[CSV/JSON] {csvp.name} / {jsonp.name}")
def dmin_ang(atoms): D = atoms.get_all_distances(mic=True); np.fill_diagonal(D, 1e9) dmin = float(D.min()); flag = "OK" if dmin>=0.60 else "⚠︎ close (<0.60Å)" print(f"[d_min] {dmin:.3f} Å → {flag}") return dmin
def bond_dist(a, i, j): # ase.geometry.get_distances の代わりに Atoms オブジェクトの get_distance メソッドを使用 return a.get_distance(i, j, mic=True)それぞれ何をしているか
snapshot(atoms, name, ...)- ASE の
plot_atomsで構造を描画し、snapshots/以下に PNG を保存します。 - 「NiO 表面の斜視図」「NiO/Pt₁₃ の真上からの図」など、記事用の図やチェック用の画像を簡単に残せます。
- ASE の
export_coords(atoms, name, fractional=False)- 原子の座標を CSV と JSON で
coords/に出力します。 fractional=Trueにすると、分率座標(fx, fy, fz)で保存されるので、「CIF と同じ座標系での確認」に便利です。
- 原子の座標を CSV と JSON で
dmin_ang(atoms)- セル内の 全ての距離行列 を計算し、その最小値 d_min を出力します。
- 0.60 Å 未満なら「原子がほぼ重なっている」と判定し、ざっくりした衝突チェックとして使います。
bond_dist(a, i, j)- ASE の
Atoms.get_distance(i, j, mic=True)を薄ラッパーしたものです。 - 周期境界を考慮した最短距離を取ってくれるので、後で Pt–C, C–O の結合距離などを確認するのに使います。
- ASE の
1. NiO(100) 表面スラブをつくる
ここでは、CO₂還元の舞台となる NiO(100) 表面スラブ を用意します。
ゴールは次の 2 つです。
- rocksalt 構造の NiO から NiO(100) スラブ を作る(
NiO100.cif) - Pt クラスターを載せやすいように、直交セル+スーパーセル に引き伸ばしたスラブ(
NiO100_rect.cif)を作る
この 2 つが、後の Pt クラスター・CO 吸着・UMA 単点計算の「土台」になります。
1-1. rocksalt NiO から NiO(100) スラブを切り出す
まずは ASE の bulk と surface を使って、NiO(100) のスラブを作ります。
格子定数は代表値として a ≈ 4.17 Å を使い、上下に十分な真空層をとっておきます。
やることは:
- rocksalt NiO バルクを作る
- (100) 面に沿って 6 層スラブを切り出し、真空 15 Å を入れる
- CIF 出力+簡易チェック(PNG・座標・最近接距離)
対応するセルはそのまま次のとおりです。
# NiO rocksalt(代表値): a≈4.17 Å
a_nio = 4.17
nio_bulk = bulk('NiO', crystalstructure='rocksalt', a=a_nio)
nio100 = surface(nio_bulk, (1,0,0), layers=6, vacuum=15.0)
nio100.center(axis=2)
write("NiO100.cif", nio100)
print("wrote NiO100.cif Natoms=", len(nio100))
snapshot(nio100, "01_NiO100_overview")
snapshot(nio100, "01_NiO100_side", rotation="90x,0y,0z")
export_coords(nio100, "01_NiO100_cart", fractional=False)
export_coords(nio100, "01_NiO100_frac", fractional=True)
dmin_ang(nio100)何をやっているか
bulk('NiO', crystalstructure='rocksalt', a=a_nio)→ NaCl 型(rocksalt)の NiO バルクを作ります。
surface(..., (1,0,0), layers=6, vacuum=15.0)→ (100) 方向に 6 層ぶん切り出し、上下に 15 Å の真空を入れたスラブを作ります。
nio100.center(axis=2)→ スラブを z 方向の中央に寄せ、上下の真空が対称になるようにしておきます。
write("NiO100.cif", ...)→ この時点の NiO(100) スラブを CIF で保存します(VESTA などで確認可能)。
snapshot(...)→ 斜視図・側面図を PNG に保存しておきます。
export_coords(...)→ 直交座標・分率座標を CSV / JSON で
coords/に書き出します。dmin_ang(nio100)→ 最近接距離 d_min を計算し、0.60 Å 未満なら「原子がほぼ重なっている」と警告してくれます。
この段階で、NiO100.cif が NiO(100) スラブの「素の状態」 として手元にあります。
1-2. 斜格子スラブを直交セル+スーパーセルに変換する
NiO100.cif の a–b 平面は、六方格子のように 60° の斜格子 になっています。
ここに Pt ナノクラスターや CO を載せていくことを考えると、
- x, y 方向の長さがそのまま距離感に対応していた方が直感的
- スーパーセル化を 整数行列 で管理しやすい
という理由から、 直交セルに変換したうえで、面内を 3×3 に広げておく 方が扱いやすくなります。
やること:
NiO100.cifを読む- 変換行列 T で「斜格子 → 直交セル」に変換
- さらに対角行列 S で面内を 3×3 に拡大
- PBC をそろえて
NiO100_rect.cifとして保存
対応するセルは次のとおりです。
from ase.io import read, write
from ase.build import make_supercell
import numpy as np
# 1) まず現在の斜格子スラブを読む
slab = read("NiO100.cif") # 斜格子っぽい a,b, z=真空 のもの
# 2) 斜格子(60°、|a|=|b|=L)→ 直交セルへ整数変換
# 変換行列 T の列が新基底ベクトルの整数係数:
# u = 2a, v = -a + 2b, w = c (u ⟂ v になり直交化できます)
T = np.array([[ 2, -1, 0], [ 0, 2, 0], [ 0, 0, 1]], dtype=int)
slab_ortho = make_supercell(slab, T)
# 3) さらに面内を拡大(Pt13がはみ出さないよう 3×3 くらいが目安)
# 直交化後の u, v が ~5–6 Å なので、3×3 で 15–18 Å 程度になります
S = np.diag([3, 3, 1]) # u×3, v×3, c×1
slab_big = make_supercell(slab_ortho, S)
# 4) PBC をすべて True(z は真空を十分に確保している前提)
slab_big.set_pbc((True, True, True))
write("NiO100_rect.cif", slab_big)
print("wrote: NiO100_rect.cif")
print("cell (Å):\n", slab_big.cell.array)変換行列 T と S のイメージ
もともとの a, b ベクトルは 60° の角度を持った斜格子です。
これを整数係数の組み合わせで
u = 2av = -a + 2bw = c
のように取り直すと、u と v がほぼ直交します。
この「新基底ベクトルの係数」を列に持つ行列が T です。
そのあと、対角行列 S = diag(3,3,1) でもう一度
make_supercellすると、u, v 方向に 3 倍ずつ広がった 3×3 スーパーセルになります。
こうして、直交セル+十分な面内サイズを持つ NiO(100) スラブ (NiO100_rect.cif)が得られます。
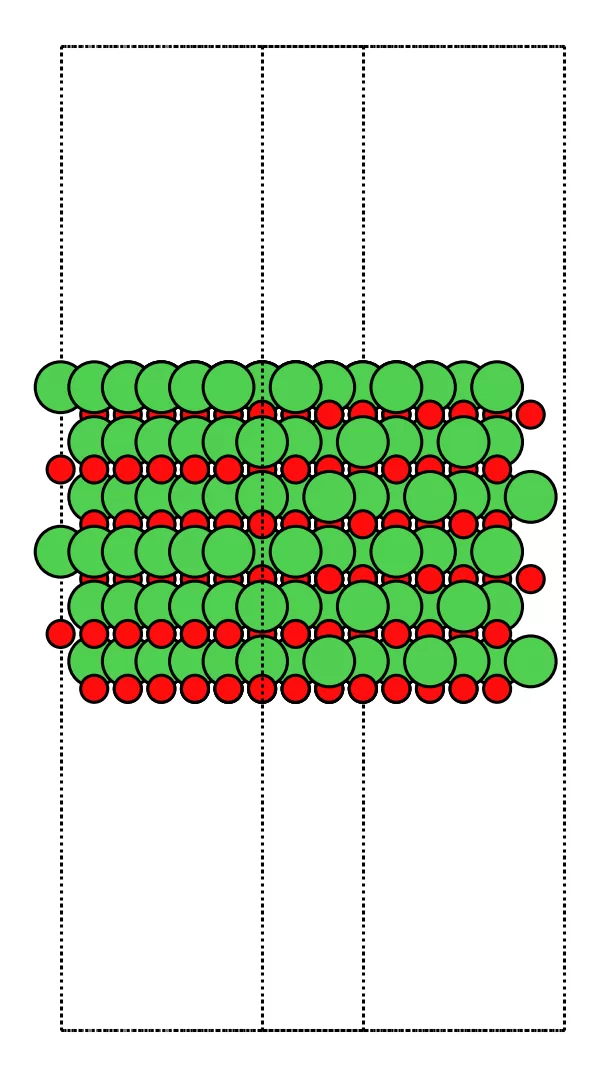
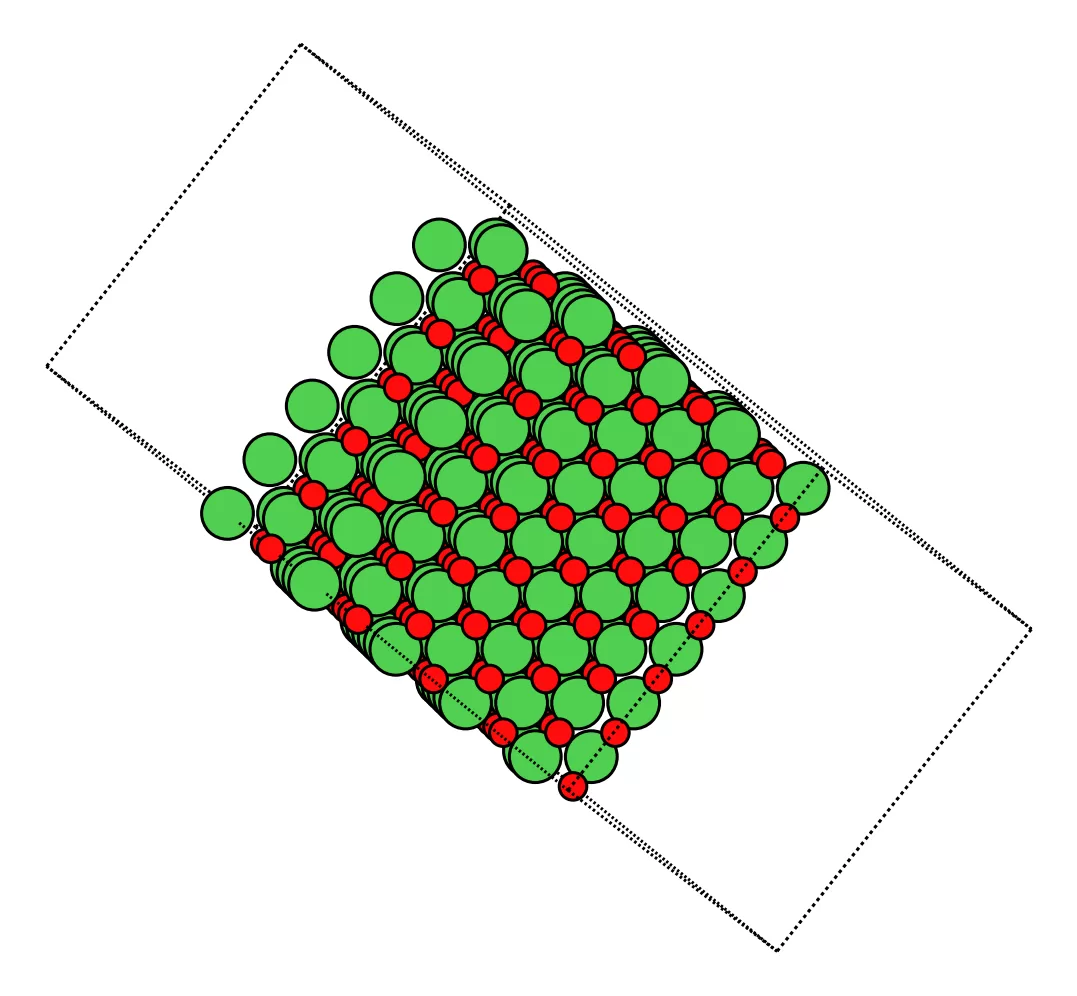
1-3. 直交化した NiO(100) スラブをざっくり可視化する
直交セル化とスーパーセル化が正しくできているか、
一度 PNG で「真上」と「斜視」の2枚を出しておくと安心です。
from ase.visualize.plot import plot_atoms
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(5,5), dpi=220)
plot_atoms(slab_big, ax, rotation='90x,0y,0z', radii=0.9, show_unit_cell=2) # 真上から
ax.set_axis_off(); plt.tight_layout(); plt.show()
fig, ax = plt.subplots(figsize=(5,5), dpi=220)
plot_atoms(slab_big, ax, rotation='25x,35y,0z', radii=0.9, show_unit_cell=2) # 斜視
ax.set_axis_off(); plt.tight_layout(); plt.show()1 枚目(真上):
表面の周期性・面内サイズがどのくらい確保できているかをチェックできます。
2 枚目(斜視):
スラブの厚みと真空層のバランスを視覚的に確認できます。
このあたりで
- 面内が 15〜18 Å 程度に広がっていそうか
- z 方向に十分な真空が入っていそうか
を確認しておくと、次章以降の Pt クラスター・CO 吸着の準備がスムーズになります。
2. NiO(100) の上に Pt₁₃ ナノクラスターを載せる
1章で NiO(100) スラブ(NiO100.cif) と
直交+拡大型の NiO100_rect.cif まで準備できました。
ここでは、いよいよ Pt₁₃ icosahedron ナノクラスター を NiO の上に載せて、
- お試し版(小さいスラブ上の Pt₁₃:
NiO100_Pt13.cif) - 本番用(直交・拡大量スラブ上の Pt₁₃:
NiO100_rect_Pt13.cif)
の 2 種類の支持体 CIF を作ります。
後の章で CO を置いたり UMA に投げたりするときに使うのは「本番用」ですが、
最初に小さいスラブ上で動かしてみると、動作チェックや高さパラメータの感覚が掴みやすくなります。
2-1. 小さな NiO(100) スラブ上に Pt₁₃ を載せてみる(お試し版)
まずは 1章で作った NiO100.cif の上に、Pt₁₃ を載せてみます。
ここでは:
Icosahedron('Pt', noshells=2)で Pt₁₃ クラスターを作成- 重心を原点に移動
- NiO(100) スラブの x–y 中央+表面 z から h Å 上 に載せる
- CIF と PNG、座標、最近接距離をざっと確認
という一連の流れを、小さいセルで試します。
# Pt13 (icosahedron) を界面上 h Å 漂わせて載せる
pt13 = Icosahedron('Pt', noshells=2) # 13 atoms
pt13.translate(-pt13.get_center_of_mass())
# x-y中央へ、zは表面最上位からhだけ上に
h = 2.4 # ← 近すぎたら 2.6/2.8 と上げる
xy_center = 0.5 * (nio100.cell[0] + nio100.cell[1])
z_top = nio100.positions[:,2].max()
pt13.translate(xy_center + np.array([0,0,z_top + h]) - pt13.get_center_of_mass())
support = nio100 + pt13
write("NiO100_Pt13.cif", support)
print("wrote NiO100_Pt13.cif Natoms=", len(support))
snapshot(support, "02_support_persp", rotation="25x,35y,0z")
snapshot(support, "02_support_top", rotation="90x,0y,0z")
export_coords(support, "02_support_cart", fractional=False)
dmin_ang(support)ここで見ておきたいポイントは 3 つです。
h の感覚
h = 2.4から始めて、Ni–Pt, O–Pt が近すぎるようなら 2.6 / 2.8… と上げていくイメージです。載り方の見た目
02_support_persp.png,02_support_top.pngの 2 枚の PNG で、「ちゃんと NiO の上に Pt の塊が載っているか」をざっくり確認します。
最近接距離 dmin
dmin_ang(support)の結果で、0.60 Å 未満になっていないかだけチェックしておくと安心です。
この NiO100_Pt13.cif は、「動作確認用の小さなモデル」として取っておきます。
本番の UMA スキャンは、次の直交・拡大量スラブ版で行います。
2-2. 直交・拡大量スラブ NiO100_rect の上に Pt₁₃ を載せる(本番用)
次に、1章で作った 直交+3×3 スーパーセル版の NiO(100) スラブ(NiO100_rect.cif)に
同様の手順で Pt₁₃ を載せ、本番用の支持体 NiO100_rect_Pt13.cif を作ります。
やることはお試し版とほぼ同じですが:
- 読み込むスラブが
NiO100_rect.cifになる - h を 2.6 Å に設定(少しだけ余裕を持たせた設定)
という違いがあります。
import numpy as np
from ase.cluster.icosahedron import Icosahedron
from ase.io import write
# 1) 最新の直交・拡大型スラブを読み込み
base = read("NiO100_rect.cif")
# 2) Pt13 生成&重心原点
pt13 = Icosahedron('Pt', noshells=2) # 13 atoms
pt13.translate(-pt13.get_center_of_mass())
# 3) 面内中央(x,y)+ 上面 z_top の h Å 上に置く
xy_center = 0.5 * (base.cell[0] + base.cell[1])
z_top = base.positions[:,2].max()
h = 2.6 # ← 近すぎたら 2.8–3.0 に上げる / 遠いなら 2.2–2.4 に下げる
pt13.translate(xy_center + np.array([0,0,z_top + h]) - pt13.get_center_of_mass())
support = base + pt13
write("NiO100_rect_Pt13.cif", support)
print("wrote: NiO100_rect_Pt13.cif")ここまでで、
NiO100_rect_Pt13.cif→ NiO(100) 直交+拡大量スラブの上に Pt₁₃ が載った「本番用支持体 CIF」
が完成します。
メモ:
直交+面内が広いスラブを使うことで、
Pt₁₃ や CO が周期画像と強く干渉するリスクを下げています。
(完全にゼロにはなりませんが、「とりあえず吸着スキャンする」には十分なサイズ感です。)
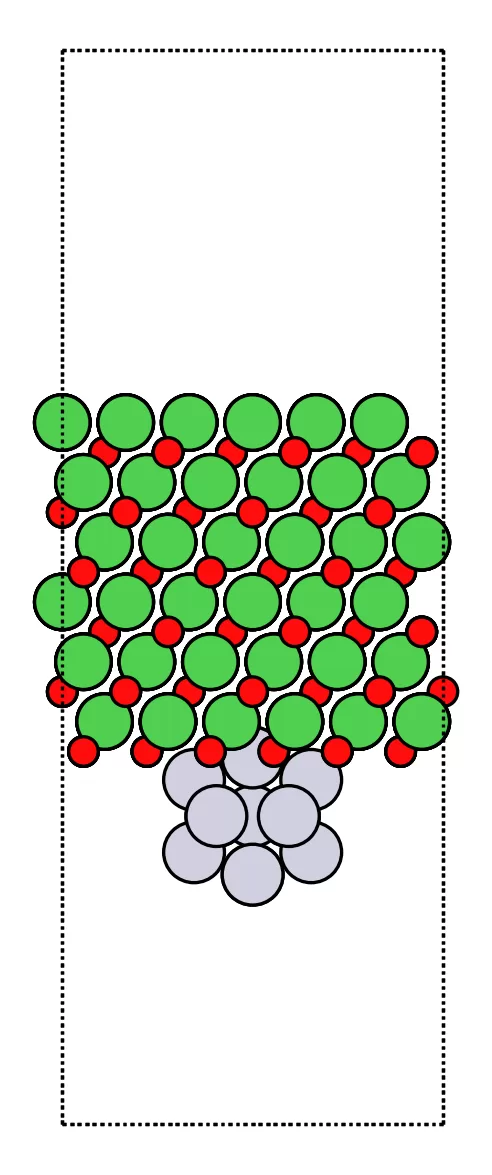
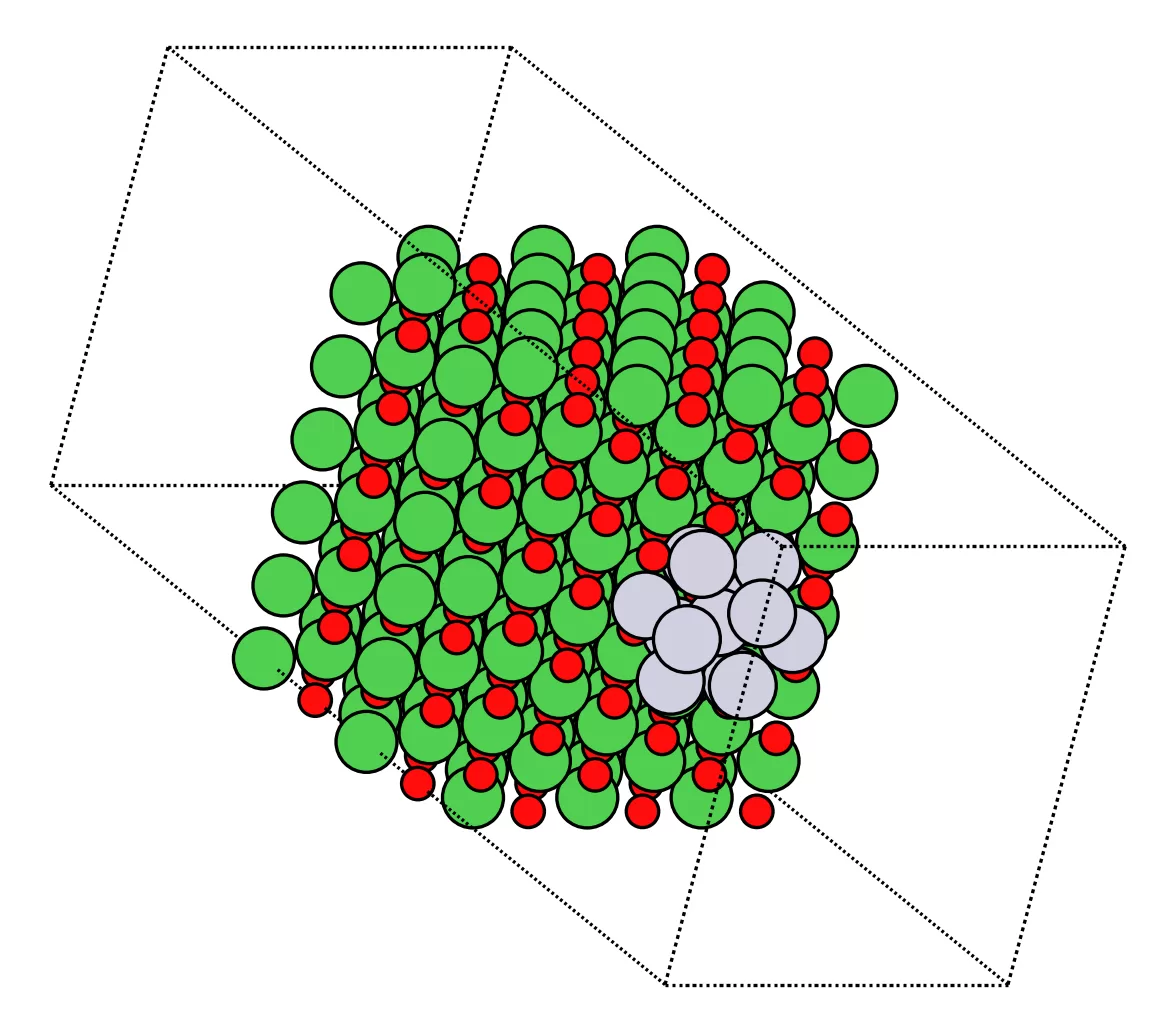
2-3. 本番用支持体 NiO100_rect_Pt13 の可視化と幾何チェック
最後に、本番用の支持体 CIF が「ちゃんと NiO の上に載っているか」「衝突していないか」を
画像と数値でざっくり確認しておきます。
# 可視化&幾何チェック(置いた後)
import numpy as np, csv
import matplotlib.pyplot as plt
from pathlib import Path
from ase.io import read
from ase.visualize.plot import plot_atoms
snap_dir = Path("snapshots"); snap_dir.mkdir(exist_ok=True)
a = read("NiO100_rect_Pt13.cif") # ← B手順で出力したファイル
# ---- 1) 画像(真上 / 斜視) ----
fig, ax = plt.subplots(figsize=(5,5), dpi=240)
plot_atoms(a, ax, rotation='90x,0y,0z', radii=0.9, show_unit_cell=2) # top view
ax.set_axis_off(); plt.tight_layout(); plt.show()
top_png = snap_dir/"B_after_place_top.png"
plt.savefig(top_png, bbox_inches="tight", pad_inches=0.02); plt.close(fig)
fig, ax = plt.subplots(figsize=(5,5), dpi=240)
plot_atoms(a, ax, rotation='25x,35y,0z', radii=0.9, show_unit_cell=2) # perspective
ax.set_axis_off(); plt.tight_layout(); plt.show()
persp_png = snap_dir/"B_after_place_persp.png"
plt.savefig(persp_png, bbox_inches="tight", pad_inches=0.02); plt.close(fig)
print(f"[PNG] {top_png}")
print(f"[PNG] {persp_png}")2 枚の PNG を見て、
- Pt クラスターが NiO の上にきちんと載っているか
- 極端に埋まっていたり、真空中に浮きすぎていないか
をざっくり確認します。
続いて、「数値的にちゃんと NiO の上にいるか」と「衝突していないか」を簡易チェックします。
# ---- 2) 上面に載っているかの数値確認 ----
z_top_surface = float(np.max([p[2] for p,sym in zip(a.positions, a.get_chemical_symbols()) if sym in ("Ni","O")]))
pt_indices = [i for i,sym in enumerate(a.get_chemical_symbols()) if sym=="Pt"]
z_pt_min = float(np.min(a.positions[pt_indices,2]))
print(f"z_top(surface) = {z_top_surface:.3f} Å")
print(f"z_min(Pt13) = {z_pt_min:.3f} Å")
print(f"Δz = z_min(Pt) - z_top(surface) = {z_pt_min - z_top_surface:.3f} Å (>=0 なら“上面”に正しく載っています)")
# ---- 3) 代表の最近接距離(衝突チェック) ----
D = a.get_all_distances(mic=True)
np.fill_diagonal(D, 1e9)
dmin = float(D.min())
flag = "OK" if dmin >= 0.60 else "⚠︎ close (<0.60 Å)"
print(f"[d_min] {dmin:.3f} Å → {flag}")Δz = z_min(Pt) - z_top(surface)→ NiO 表面の一番上の原子と、一番下の Pt の高さ差
→ これが 0 以上なら、「Pt クラスター全体が NiO より上にある」ことが保証されます。
dmin→ セル内で一番短い原子間距離(周期境界を考慮)
→ 0.60 Å 未満なら「どこかでほぼ重なっている」ので、h の見直しなどを検討
最後に、「上面付近にはどんな原子がいるのか」を z 座標でソートしてリスト化しておきます。
# ---- 4) 上面近傍の原子を z 高い順で確認(視覚と数値の対応づけ) ----
idx_sorted = np.argsort(a.positions[:,2])[::-1]
rows = []
for i in idx_sorted[:25]: sym = a[i].symbol x,y,z = a.positions[i] rows.append((i, sym, x, y, z)) print(f"idx={i:3d} {sym:>2s} x={x:8.3f} y={y:8.3f} z={z:8.3f}")
# CSV保存(任意)
import csv, os
with open("coords/B_after_place_top25.csv","w",newline="") as f: w=csv.writer(f); w.writerow(["index","symbol","x","y","z(Å)"]); w.writerows(rows)
print("[CSV] coords/B_after_place_top25.csv")この B_after_place_top25.csv を見ておくと、
- 「一番上の Pt がどの index か」
- 「その周りの Ni/O はどれか」
を後から落ち着いて追えるようになります。
次の章で CO を置くときに、「どの Pt を『頂上サイト』として扱うか」を決める材料にもなります。
3. CO 吸着候補を幾何条件でふるいにかける
1–2章で NiO(100) 直交スラブ+Pt₁₃ クラスター まで準備できました。
ここからはいよいよ CO を載せていきますが、いきなり UMA で全パターンを計算するのは少しもったいないので、
いったん「幾何条件だけ」で
衝突している構造・明らかにおかしい構造を落としておく
という前処理を挟みます。
この章では、
事前に作っておいた CO 吸着候補 CIF 群(
cands) を読み込む各構造について
セル内の最近接距離 d_min
一番上の Pt と C の距離(Pt–C)
C–O 結合長(C–O)
を計算する
しきい値(d_min / Pt–C)でフィルタして、「幾何的にマトモな候補」だけのリスト を作る
上位 12 構造のサムネイル PNG を出して、ざっと目視確認する
ところまでを行います。
💡 前提:
ここで使う
candsは、「NiO100_rect_Pt13 の上に CO をいろいろな向き・位置で置いて CIF に書き出したもの」のパスリストを想定しています。それを別セルで
cands = sorted(glob.glob("NiO_Pt13_CO_*.cif"))のように用意しておきます。
3-1. 幾何情報(d_min, Pt–C, C–O)を一括で抜き出す
まずは、候補構造ごとに
- 全原子間距離の最小値 d_min(衝突チェック用)
- 一番上の Pt と CO の C の距離(Pt–C)
- CO 分子の C–O 距離(C–O)
の 3 つを計算し、行リストにまとめます。
from ase.io import read
from pathlib import Path
from ase.geometry import get_distances # get_distancesは個別のatomsに対して使うためインポートを修正
def mic_dist(a, i, j): # atomsオブジェクトのget_distanceメソッドをmic=Trueで使う return a.get_distance(i, j, mic=True)
def dmin_ang(a): D = a.get_all_distances(mic=True) import numpy as np np.fill_diagonal(D, 1e9) return float(D.min())
rows=[]
for p in sorted(cands): a = read(p) pt = max([i for i,x in enumerate(a) if x.symbol=="Pt"], key=lambda i: a.positions[i,2]) c = [i for i,x in enumerate(a) if x.symbol=="C"][-1] o = [i for i,x in enumerate(a) if x.symbol=="O"][-1] rows.append((Path(p).name, dmin_ang(a), mic_dist(a,pt,c), mic_dist(a,c,o)))何をしているか
mic_dist(a, i, j)→ ASE
Atoms.get_distance(i, j, mic=True)を薄ラッパーしたもの。周期境界(minimum image convention)込みで距離を取ります。
dmin_ang(a)→
a.get_all_distances(mic=True)で全ペアの距離行列を取り、対角成分を巨大値にしてから最小値 d_min を抜き出しています。
「どこかで原子同士が重なっていないか?」のざっくりチェック用です。
pt:→
z座標が最大の Pt(=一番上にいる Pt)を選んでいます。CO を「頂上の Pt」に吸着させた候補を想定。
c,o:→ C と O は最後に出てくるインデックス(=吸着 CO の原子)を取る前提で、
C–O 結合長を取っています。
こうして rows には
(file_name, dmin_A, PtC_A, CO_A)のタプルが候補構造の数だけ溜まっていきます。
3-2. しきい値でフィルタして「幾何的にマトモな候補」だけを残す
次に、この rows を元に
- 衝突している構造(d_min < 0.60 Å)
- Pt–C が明らかにおかしい構造(短すぎ / 長すぎ)
を落としておきます。ここでは代表値として、
- d_min ≥ 0.60 Å
- 1.60 Å ≤ Pt–C ≤ 2.20 Å
をしきい値にしています。
# フィルタ(衝突と過度な離隔を除外)
keep = [r for r in rows if r[1] >= 0.60 and 1.60 <= r[2] <= 2.20] # dmin>=0.60Å 且つ Pt–C∈[1.6,2.2]Å
keep.sort(key=lambda x: (x[2], x[1])) # Pt–Cが短い順にざっくりここでの考え方:
d_min ≥ 0.60 Å
→ 「原子がほぼ重なっている構造」を弾くためのごくラフな条件です。
(本当に厳密にやりたい場合は、元素組ごとにしきい値を変えるなどの工夫もあり得ます。)
Pt–C ≈ 1.8 Å 前後が典型的なオンサイト吸着距離なので、
ざっくり 1.6〜2.2 Å の間にあるものだけ残しています。
keep を Pt–C が短い順(=結合が強そうな順)+ d_min の大きさでソートしておくと、
あとで「良さそうな候補」から順に UMA スキャンをかけるときに便利です。
3-3. CSV にまとめて、上位候補のサムネイルを作る
最後に、
- すべての候補の幾何情報(
CO_candidates_geom.csv) - フィルタ後の「幾何的にマトモな候補」だけのリスト(
CO_candidates_filtered.csv)
を CSV に出力し、さらに上位 12 構造のサムネイル PNG を保存します。
# 保存とサムネ(上位12件)
import csv, os
os.makedirs("coords", exist_ok=True); os.makedirs("snapshots", exist_ok=True)
with open("coords/CO_candidates_geom.csv","w",newline="") as f: w=csv.writer(f); w.writerow(["file","dmin_A","PtC_A","CO_A"]); w.writerows(rows)
with open("coords/CO_candidates_filtered.csv","w",newline="") as f: w=csv.writer(f); w.writerow(["file","dmin_A","PtC_A","CO_A"]); w.writerows(keep)
print(f"kept {len(keep)} / {len(rows)} candidates → coords/CO_candidates_filtered.csv")
# サムネ保存
for name,_,_,_ in keep[:12]: a = read(name) fig, ax = plt.subplots(figsize=(4.2,4.0), dpi=220) plot_atoms(a, ax, rotation='30x,25y,0z', radii=0.9, show_unit_cell=2) ax.set_axis_off(); plt.tight_layout() plt.savefig(f"snapshots/{Path(name).stem}.png", bbox_inches="tight", pad_inches=0.02) plt.close(fig)
print("[PNG] snapshots/cand_... (上位12枚)")できあがるファイル
coords/CO_candidates_geom.csv→ 全候補について
(ファイル名, d_min, Pt–C, C–O)をまとめた表。しきい値を変えたくなったときは、この CSV から再フィルタできます。
coords/CO_candidates_filtered.csv→ 幾何条件を満たした候補のみのリスト。
次章以降で UMA 計算に回すのは基本的にこちら。
snapshots/○○.png(最大 12 枚)→ 「見た目がどうなっているか」をサクッと確認するためのサムネイル。
CO の向き(直立/傾き/ブリッジっぽい配置など)や、Pt₁₃ のどのサイトに 乗っているかを直感的に把握できます。
4. UMA で NiO/Pt₁₃/CO の吸着エネルギーを一括スキャンする
ここまでで、
- NiO(100) の直交・拡大型スラブ
NiO100_rect.cif - その上に Pt₁₃ クラスターを載せた支持体
NiO100_rect_Pt13.cif - 幾何チェックを通過した CO 吸着候補のリスト
keep
がそろいました。
4章では、いよいよ 機械学習ポテンシャル UMA を使って、
- 各候補構造のエネルギーを計算し
- 吸着エネルギー
Eadsを求め - 「もっとも安定な CO 吸着構造」を 1 つ選び出す
ところまで進みます。
4-1. UMA ユニットを準備して参照エネルギーを計算する
最初に、ASE から呼び出せる形で UMA を用意し、 次の 2 つの参照エネルギーを計算します。
- NiO/Pt₁₃ 支持体だけのエネルギー
E_support - 気相 CO 分子のエネルギー
E_CO
記事の頭で !pip install と huggingface-cli login はすでに済んでいる前提です。
# 事前に: !pip -q install fairchem-core ; !huggingface-cli login -y
from fairchem.core import FAIRChemCalculator, pretrained_mlip
from ase.build import molecule
# UMAユニット
unit = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(unit, task_name="oc20") # 触媒系の一般タスク例
# 参照エネルギー(支持体&孤立CO)
support = read("NiO100_rect_Pt13.cif"); support.calc = calc
co_free = molecule("CO"); co_free.set_cell([20,20,20]); co_free.set_pbc([True]*3); co_free.center(); co_free.calc = calc
E_support = support.get_potential_energy(); E_co = co_free.get_potential_energy()
print(f"E_support={E_support:.3f} eV, E_CO={E_co:.3f} eV")ここでやっていることを整理すると:
get_predict_unit("uma-s-1p1")→ UMA の事前学習済みモデルを 1 つ選んで取得しています。FAIRChemCalculator(...)→ このユニットを ASE の Calculator としてラップし、Atoms.calcにセットできる形にしています。support(NiO/Pt₁₃)とco_free(孤立 CO)にcalcをセットし、get_potential_energy()を呼ぶだけで、それぞれのエネルギーが計算されます。
この 2 つの値を、あとの吸着エネルギー計算で「引き算の基準」として使います。
吸着エネルギーの定義(この章ではずっとこの約束を使います)
Etot… ある候補構造(NiO/Pt₁₃/CO 全体)のエネルギーE_support… NiO/Pt₁₃ 支持体だけのエネルギーE_CO… 気相 CO のエネルギーとすると、吸着エネルギー
Eadsは
Eads = Etot - E_support - E_COと定義しています。 このとき、
Eadsが 0 より 負の値になるほど、吸着が有利(発熱的) という解釈になります。
4-2. フィルタ済み候補の吸着エネルギーをまとめて評価する
次に、3章で幾何フィルタを通過した候補リスト keep について、
UMA で単点計算を実行し、それぞれの Eads を求めていきます。
import time
results=[]
for name,_,_,_ in keep: a = read(name); a.calc = calc Etot = a.get_potential_energy() eads = Etot - E_support - E_co results.append((name, eads)) print(f"{name:45s} Eads = {eads:7.3f} eV")
results.sort(key=lambda x: x[1])
with open("coords/Eads_scan_UMA.csv","w",newline="") as f: w=csv.writer(f); w.writerow(["file","Eads_eV"]); w.writerows(results)
print("\\nTop-5 (most stable):")
for r in results[:5]: print(r)
best_name, best_e = results[0]
print(f"\\nBEST = {best_name} ({best_e:.3f} eV)")ここでのポイントは次の通りです。
for name,_,_,_ in keep:→keepは(file, dmin_A, PtC_A, CO_A)というタプルのリストでしたが、 ここではファイル名nameだけを使うので、残りは_で捨てています。- 毎回
a.calc = calcとして UMA をセットし、get_potential_energy()でEtotを計算。 - 吸着エネルギーは、先ほどの約束どおり
Eads = Etot - E_support - E_coで計算しています。 results.sort(key=lambda x: x[1])→resultsは(ファイル名, Eads)のリストなので、x[1](吸着エネルギー)が小さい順、つまり「安定な順」に並び替えています。Eads_scan_UMA.csv→ すべての候補について(ファイル名, Eads)を保存した CSV です。 後から別のしきい値で選び直したり、他の指標と組み合わせて解析したりするときに便利です。
最後に、
best_name… 最もEadsが小さかった構造のファイル名best_e… そのときのEadsの値
を拾って、ログに「BEST = …」と出力しています。
4-3. 最安定構造を CIF と PNG にして取り出す
計算の結果、もっとも吸着エネルギーが安定だった構造を最終的な CIF と図として残す セルがこちらです。
best = read(best_name)
write("FINAL_NiO100_Pt13_CO.cif", best)
print("wrote FINAL_NiO100_Pt13_CO.cif")
# 可視化
fig, ax = plt.subplots(figsize=(5,4.6), dpi=240)
plot_atoms(best, ax, rotation='30x,25y,0z', radii=0.9, show_unit_cell=2)
ax.set_axis_off(); plt.tight_layout(); plt.show()
plt.savefig("snapshots/FINAL_persp.png", bbox_inches="tight", pad_inches=0.02); plt.close(fig)
fig, ax = plt.subplots(figsize=(5,4.6), dpi=240)
plot_atoms(best, ax, rotation='90x,0y,0z', radii=0.9, show_unit_cell=2)
ax.set_axis_off(); plt.tight_layout(); plt.show()
plt.savefig("snapshots/FINAL_top.png", bbox_inches="tight", pad_inches=0.02); plt.close(fig)
print("[PNG] snapshots/FINAL_persp.png / FINAL_top.png")
# 代表距離(Pt–C, C–O)
pt = max([i for i,x in enumerate(best) if x.symbol=="Pt"], key=lambda i: best.positions[i,2])
c = [i for i,x in enumerate(best) if x.symbol=="C"][-1]
o = [i for i,x in enumerate(best) if x.symbol=="O"][-1]
# def mic_dist(a, i, j):
# return float(get_distances(a.positions[i], a.positions[j:j+1],
# cell=a.cell, pbc=a.pbc, mic=True)[1][0][0])
# print(f"Pt–C = {mic_dist(best,pt,c):.3f} Å, C–O = {mic_dist(best,c,o):.3f} Å")
# print(f"Eads(best, UMA) = {best_e:.3f} eV (negative=favorable)")このセルで出てくる成果物は:
FINAL_NiO100_Pt13_CO.cif→ NiO(100) 上の Pt₁₃ クラスターに CO が吸着した「最安定候補」の CIF。 VESTA や Avogadro に渡して、構造を詳細に確認したり、 そのまま別の計算コード(DFT など)の初期構造として使ったりできます。
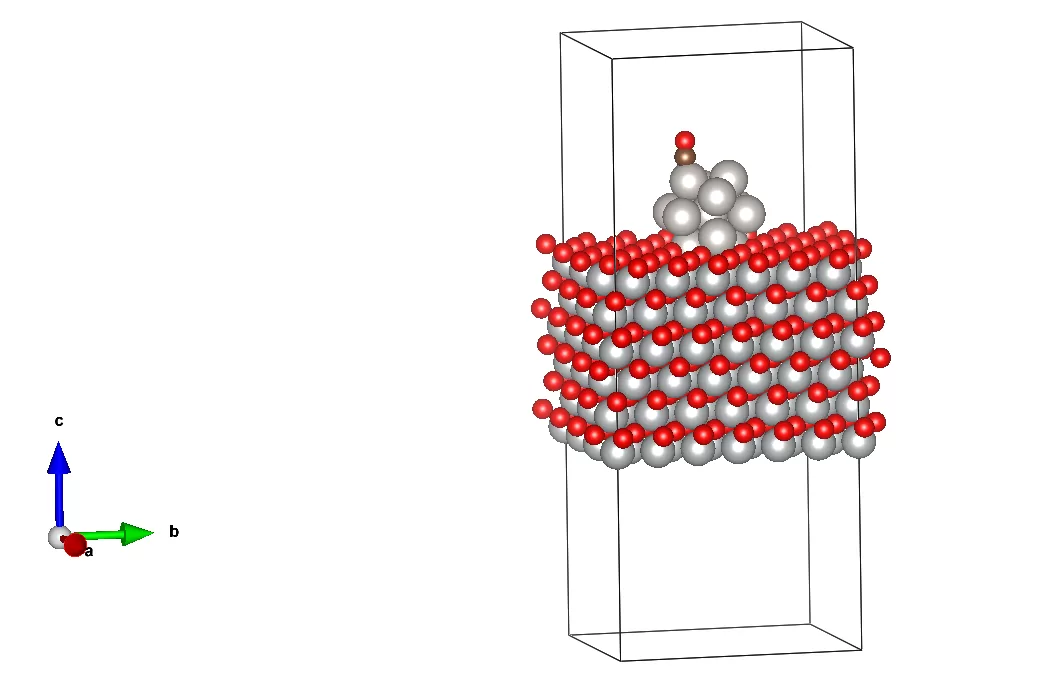
FINAL_persp.png,FINAL_top.png→ それぞれ「斜めから見た図」「真上から見た図」です。 どの Pt サイトに CO が乗っているか、CO がどの向きか、といった情報を直感的に把握できます。
コメントアウトされている部分を有効にすると、
- 一番上の Pt と CO の C の距離(Pt–C)
- CO の結合長(C–O)
- 最安定構造の吸着エネルギー
Eads(best)
も一緒に出力できます。 例えば、
Pt–C ≒ 1.8 ÅC–O ≒ 1.15 ÅEads ≒ -x.x eV
といったオーダーであれば、「CO 吸着としてそれらしいサイズ感か?」を 文献値とざっくり比較することもできます。
5. 他の系に応用するときの「ユースケース別ガイド」
ここからは、次の 3 パターンに分けて整理します。
- 分子だけ変えたい:NiO + Pt₁₃ はそのまま、CO → CO₂ / H₂O など
- クラスターだけ変えたい:NiO(100) はそのまま、Pt₁₃ → Au₁₃ / Au₅₅ など
- 表面(支持体)から変えたい:NiO(100) → 別の酸化物・金属表面(外部 CIF 含む)
それぞれ「このユースケースなら、ノートのどこを変えればいいか」だけをまとめます。
5-1. ユースケース①:分子だけ変えたい(CO → CO₂ / H₂O など)
NiO(100) + Pt₁₃ はそのまま で、
「CO じゃなくて CO₂ を吸着させてみたい」みたいなケース。
変える場所はざっくりこの 3 つだけ
- 孤立分子の生成(参照エネルギー用)
- 候補構造の生成スクリプト(
candsを作っているセル) - インデックスの取り方(C と O をどう拾うか)
① 孤立分子の生成
今は 4 章でこうなっていました:
from ase.build import molecule
co_free = molecule("CO")
co_free.set_cell([20,20,20])
co_free.set_pbc([True]*3)
co_free.center()
co_free.calc = calc
E_co = co_free.get_potential_energy()ここを、例えば CO₂ にするなら:
co2_free = molecule("CO2")
co2_free.set_cell([20,20,20])
co2_free.set_pbc([True]*3)
co2_free.center()
co2_free.calc = calc
E_co2 = co2_free.get_potential_energy()に変えるだけです。
あとは 4 章の吸着エネルギー定義を
Eads = Etot - E_support - E_CO2のように、変数名を合わせてあげれば OK です。
② 吸着候補 cands の作り方
ここはノートの外(あなたが別セルで書いているはずの部分)ですが、考え方は同じで、
- CO の代わりに CO₂ を載せる
- 高さ h と向き(どちらが表か、どれくらい寝かせるか)を変えながら CIF を量産する
- その CIF のパスリストを
candsに入れる
という流れはまったく同じです。
たとえば、
- C が金属に近づいている「エンドオン」型
- O–C–O 全体が寝ている「ブリッジ」型
の 2 パターンを for ループで作っておく、みたいなイメージです。
③ 幾何フィルタのインデックスまわり
3 章のこの部分:
pt = max([i for i,x in enumerate(a) if x.symbol=="Pt"], key=lambda i: a.positions[i,2])
c = [i for i,x in enumerate(a) if x.symbol=="C"][-1]
o = [i for i,x in enumerate(a) if x.symbol=="O"][-1]
rows.append((Path(p).name, dmin_ang(a), mic_dist(a,pt,c), mic_dist(a,c,o)))CO₂ に変えた場合も、
- 「一番上の Pt」はそのまま
- C と O の扱いだけ少し考える
だけです。
単純に「最後の C と最後の O」を使うやり方でも動きますが、
- C だけ使いたいなら
c_indexだけあれば良い - 分子内 C–O をチェックしたい場合は、CO₂ の 2 本分を全部見る or 代表値だけ見る、など
少しロジックを足すイメージです。
5-2. ユースケース②:クラスターだけ変えたい(Pt₁₃ → Au₁₃ / Au₅₅ など)
NiO(100) はそのまま、金属クラスターだけ変えたいケース。
変える場所はこの 3 つ
- クラスター生成の行(元素とサイズ)
- 載せる高さ
h - (必要なら)スーパーセルのサイズ
① クラスター生成
2 章でやっていたここです:
from ase.cluster.icosahedron import Icosahedron
pt13 = Icosahedron('Pt', noshells=2)
pt13.translate(-pt13.get_center_of_mass())これを例えば Au₁₃ にするなら:
au13 = Icosahedron('Au', noshells=2)
au13.translate(-au13.get_center_of_mass())Au₅₅ にするなら:
au55 = Icosahedron('Au', noshells=3) # noshells=3 → 55 atoms
au55.translate(-au55.get_center_of_mass())に変えるだけです。
あとは変数名 pt13 → au13 に合わせて、
support = base + pt13 のところを support = base + au13 のように変えれば OK。
② 支持体との距離 h
同じセルにあるこの行:
h = 2.6 # ← 近すぎたら 2.8–3.0 に上げる / 遠いなら 2.2–2.4 に下げるは、元素やクラスターサイズによって微調整ポイントです。
- まずは 2.5〜3.0 Å あたりから始める
- 2 章の
dminチェックで「近すぎないか」を確認する - PNG を見て、明らかに埋まっていないか/離れすぎていないかを見る
という流れに変わりません。
③ スーパーセルサイズ(必要なら)
クラスターを大きく(Au₅₅ など)したときに、
- クラスター同士が周期境界で近づきすぎていそう
- 図を見て「明らかに隣と干渉している」
となったら、1 章のここ:
S = np.diag([3, 3, 1]) # u×3, v×3, c×1を np.diag([4,4,1]) のように変えて、面内を広げてください。
5-3. ユースケース③:表面(支持体)から変えたい
最後は一番大きく変えるパターン、
NiO(100) 自体を別の表面に変えたい ケースです。
これはさらに 2 パターンに分かれます:
- ③-A:ASE の
bulk/surfaceで作れる単純な結晶 - ③-B:COD / Materials Project などから CIF をダウンロードして使う
③-A:ASE だけで完結させる場合
1 章のこの部分を中心に変えます:
a_nio = 4.17
nio_bulk = bulk('NiO', crystalstructure='rocksalt', a=a_nio)
nio100 = surface(nio_bulk, (1,0,0), layers=6, vacuum=15.0)例えば Cu(111) 表面にしたい場合は:
cu_bulk = bulk('Cu', crystalstructure='fcc', a=a_cu) # a_cu は文献値を入れる
cu111 = surface(cu_bulk, (1,1,1), layers=6, vacuum=15.0)のようになります。
その後の流れ(center(axis=2), CIF 出力、直交化、スーパーセル化)は
NiO のときと全く同じです。
③-B:外部 CIF から始める場合
NiO のときは bulk → surface でしたが、外部 CIF を使う場合は:
- ダウンロードした CIF をそのまま
read("xxx.cif")する - 必要なら
SlabGeneratorなどで表面切り出し - その結果を
NiO100_rect.cif相当のファイル名にしてしまう
という運用にしてしまうのが楽です。
例えば:
slab = read("MySurface_from_COD.cif")
write("NiO100_rect.cif", slab)としてしまえば、以降のコード(Pt₁₃ を載せる部分)は
そのまま動きます(ファイル名に NiO と書いてあっても中身は別物でも良い、という割り切り)。
きれいにやりたければ、ファイル名を
CeO2_111_rect.cifCeO2_111_rect_Au13.cif
のように全部変えていく、という感じです。
5-4. まとめ:ユースケースごとに「変える場所」を決める
もう一度だけ整理すると:
- 分子だけ変えたい(CO → CO₂ など)
- 孤立分子の生成(4 章の
molecule("CO")の部分) - 吸着候補
candsを作っているスクリプト - 3 章の C/O インデックス取得と、距離の閾値
- 孤立分子の生成(4 章の
- クラスターだけ変えたい(Pt₁₃ → Au₁₃ / Au₅₅ など)
Icosahedron('Pt', noshells=2)の行(元素と殻数)- 支持体との距離
h - 必要ならスーパーセルサイズ(
S = np.diag([...]))
- 表面から変えたい(NiO → 別の表面)
- 1 章の
bulk/surface部分、または外部 CIF の読み込み - 場合によっては直交化の行列
TとスーパーセルS - 出力ファイル名(
NiO100_rect.cifなど)を系に合わせて変更
- 1 章の
最後に
いかがでしたか?本記事で紹介した分子構造の作り方を一度試していただき、機械学習ポテンシャルを使って計算を回していただくと、計算化学がより身近に感じられると思います。Cifファイルは論文などによく記載されているので、金属種を変えて計算するだけでも面白いと思いますので是非試してみてください。
参考文献
- K. Smith et al. “UMA: Universal Machine-learning Interatomic Potential,” arXiv preprint, arXiv:2505.08762 (2025). <https://arxiv.org/abs/2505.08762>
- B. M. Wood et al., “UMA: A Family of Universal Models for Atoms,” arXiv preprint, arXiv:2506.23971 (2025). <https://arxiv.org/abs/2506.23971>
- FAIRchem Team, “FAIRChem: FAIR Chemistry Toolkit,” GitHub Repository (accessed 2025-11-24). <https://github.com/facebookresearch/fairchem>
- Meta FAIR Chemistry, “facebook/UMA,” Hugging Face Model Card (accessed 2025-11-24). <https://huggingface.co/facebook/UMA>
- A. H. Larsen et al., “The atomic simulation environment—a Python library for working with atoms,” J. Phys.: Condens. Matter 29, 273002 (2017). https://doi.org/10.1088/1361-648X/aa680e
- S. P. Ong et al., “Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis,” Comput. Mater. Sci. 68, 314–319 (2013). https://doi.org/10.1016/j.commatsci.2012.10.028
- G. Landrum, “RDKit: Open-Source Cheminformatics Software,” RDKit Project (accessed 2025-11-24). <https://www.rdkit.org>
- K. Momma and F. Izumi, “VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data,” J. Appl. Crystallogr. 44, 1272–1276 (2011). https://doi.org/10.1107/S0021889811038970
- M. D. Hanwell et al., “Avogadro: an advanced semantic chemical editor, visualization, and analysis platform,” J. Cheminf. 4, 17 (2012). https://doi.org/10.1186/1758-2946-4-17
- International Union of Crystallography, “checkCIF/PLATON: Validation of crystallographic information files,” IUCr checkCIF service (accessed 2025-11-24). <https://checkcif.iucr.org>
- K. T. Winther et al., “Catalysis-Hub.org, an open electronic structure database for surface reactions,” Sci. Data 6, 75 (2019). https://doi.org/10.1038/s41597-019-0081-y
- L. Chanussot et al., “The Open Catalyst 2020 (OC20) dataset and community challenges,” ACS Catal. 11, 6059–6072 (2021). https://doi.org/10.1021/acscatal.0c04525
- A. Sápi et al., “In Situ DRIFTS and NAP-XPS Exploration of the Complexity of CO₂ Hydrogenation over Size-Controlled Pt Nanoparticles Supported on Mesoporous NiO,” J. Phys. Chem. C 122(10), 5553–5565 (2018). https://doi.org/10.1021/acs.jpcc.8b00061
- Meta FAIR Chemistry & collaborators, “Open Molecules 2025 (OMol25) Dataset, Evaluations, and Models,” arXiv preprint, arXiv:2505.08762 (2025) and related resources (overview in Meta FAIR news, accessed 2025-11-24).
Gaussianを使った量子化学計算の初心者向け技術書を販売中
しばしば出くわすエラーへの対処法をはじめ
Gaussianと無料ソフトウェア Avogadro を組み合わせた物性解析手法が学べます!

