

本記事では、プログラミング言語Rを利用した、大規模がんゲノムプロジェクト The Cancer Genome Atlas(TCGA)のRNA-Seqデータ及び臨床データの取得・解析の手順を紹介しています。
macOS Sequoia (15.5)


公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!

公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!


TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます
概要
ここでは、TCGAの中でも代表的ながん種である 乳がん(TCGA-BRCA) を対象とします。
解析には、Bioconductorで提供されているcuratedTCGADataとTCGAutilsパッケージを使用します。curatedTCGADataはTCGAの各種オミックスデータ(遺伝子発現、変異、コピー数、メチル化、miRNA、RPPA など)を、各がん種ごとに 統一された形式(MultiAssayExperiment) で取得できるパッケージです。
データは Broad Genome Data Analysis Center(Broad GDAC) に由来し、整形済みで即解析可能です。
MultiAssayExperimentに限らず、Bioconductorにある「~Experiment」という名前のパッケージ(例: SingleCellExperiment, SummarizedExperiment)は、同名のオブジェクトを定義するようなパッケージであり、オミックスの計測データ(例: 発現量行列)やメタデータ(例: 実験条件、性別)を1まとめにして、それを操作するための関数も統一することで、データが新しくなる度に、ユーザーが独自に前処理をする手間を省いてくれます。
参考: https://rpubs.com/mramos/curatedTCGAWorkshop
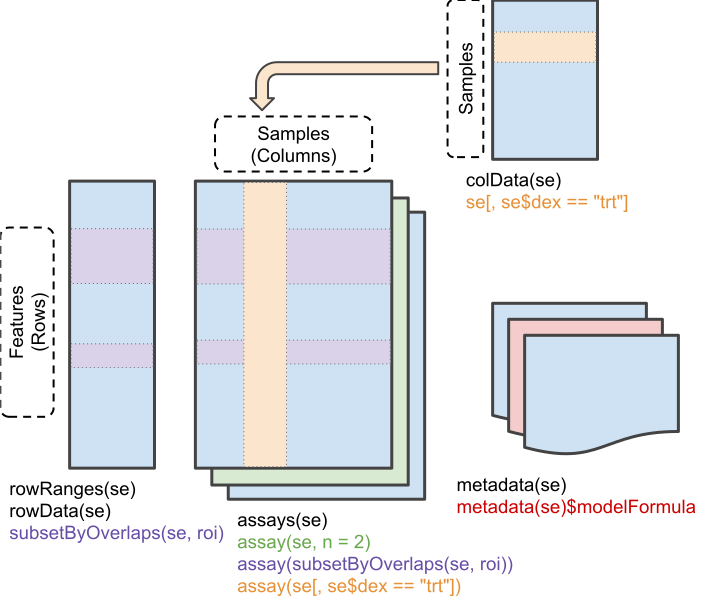
TCGAutilsはTCGA特有のバーコード形式やサンプル種別の管理、複数データの整合などを支援するためのユーティリティパッケージで、curatedTCGADataと組み合わせることで、サンプルのフィルタやIDマッピングなどが容易に行えます。
curatedTCGAData / TCGAutilsのインストール
R言語やR-studio(またはPositron)は事前にインストールされているものとします。
各OSにインストールされたR(WindowsならR.exe, MacならR.app)のコンソール画面か、もしくはR-Studio/Positronを起動し、以下のコマンドを入力することで、curatedTCGADataやTCGAutilsの内部で実装された関数や、内部データを利用できるようになります。
# curatedTCGAData / TCGAutilsがない場合は、インストール
if (!require("curatedTCGAData", quietly = TRUE)) install.packages("curatedTCGAData")
if (!require("TCGAutils", quietly = TRUE)) install.packages("TCGAutils")
# パッケージをロード
library(curatedTCGAData)
library(TCGAutils)TCGAデータの取得
まず、curatedTCGADataパッケージに含まれている全33種類のがん種略称を確認します。
data("diseaseCodes")
knitr::kable(diseaseCodes, align = "l", escape = FALSE, caption = "List of available cancer types from curatedTCGAData") %>% kableExtra::kable_styling( bootstrap_options = c("hover", "striped", "responsive"), full_width = FALSE )
ここでは、その中から 乳がん(BRCA) を対象とします。
> curatedTCGAData("BRCA", version="2.1.1")
#snapshotDate(): 2025-04-12
#See '?curatedTCGAData' for 'diseaseCode' and 'assays' inputs
#Querying EH with: BRCA_CNASeq-20160128
#Querying EH with: BRCA_CNASNP-20160128
#Querying EH with: BRCA_CNVSNP-20160128
#Querying EH with: BRCA_GISTIC_AllByGene-20160128
#Querying EH with: BRCA_GISTIC_Peaks-20160128
#Querying EH with: BRCA_GISTIC_ThresholdedByGene-20160128
#Querying EH with: BRCA_Methylation_methyl27-20160128_assays
#Querying EH with: BRCA_Methylation_methyl27-20160128_se
#Querying EH with: BRCA_Methylation_methyl450-20160128_assays
#Querying EH with: BRCA_Methylation_methyl450-20160128_se
#Querying EH with: BRCA_mRNAArray-20160128
#Querying EH with: BRCA_Mutation-20160128
#Querying EH with: BRCA_RNASeq2Gene-20160128
#Querying EH with: BRCA_RNASeqGene-20160128
#Querying EH with: BRCA_RPPAArray-20160128
#Querying EH with: BRCA_miRNASeqGene-20160128
#Querying EH with: BRCA_RNASeq2GeneNorm-20160128
# ah_id title file_size
#1 EH4769 BRCA_CNASeq-20160128 0 Mb
#2 EH4770 BRCA_CNASNP-20160128 9.8 Mb
#3 EH4771 BRCA_CNVSNP-20160128 2.8 Mb
#4 EH4773 BRCA_GISTIC_AllByGene-20160128 1.2 Mb
#5 EH4774 BRCA_GISTIC_Peaks-20160128 0 Mb
#6 EH4775 BRCA_GISTIC_ThresholdedByGene-20160128 0.3 Mb
#7 EH4777 BRCA_Methylation_methyl27-20160128_assays 60.7 Mb
#8 EH4778 BRCA_Methylation_methyl27-20160128_se 0.4 Mb
#9 EH4779 BRCA_Methylation_methyl450-20160128_assays 2646.4 Mb
#10 EH4780 BRCA_Methylation_methyl450-20160128_se 6 Mb
#11 EH4782 BRCA_mRNAArray-20160128 27.3 Mb
#12 EH4783 BRCA_Mutation-20160128 4.5 Mb
#13 EH4784 BRCA_RNASeq2Gene-20160128 43.1 Mb
#14 EH4786 BRCA_RNASeqGene-20160128 30 Mb
#15 EH4787 BRCA_RPPAArray-20160128 1.5 Mb
#16 EH8124 BRCA_miRNASeqGene-20160128 2 Mb
#17 EH8125 BRCA_RNASeq2GeneNorm-20160128 93.6 Mb
# rdataclass rdatadateadded rdatadateremoved
#1 RaggedExperiment 2021-01-27 <NA>
#2 RaggedExperiment 2021-01-27 <NA>
#3 RaggedExperiment 2021-01-27 <NA>
#4 SummarizedExperiment 2021-01-27 <NA>
#5 RangedSummarizedExperiment 2021-01-27 <NA>
#6 SummarizedExperiment 2021-01-27 <NA>
#7 SummarizedExperiment 2021-01-27 <NA>
#8 SummarizedExperiment 2021-01-27 <NA>
#9 RaggedExperiment 2021-01-27 <NA>
#10 SummarizedExperiment 2021-01-27 <NA>
#11 SummarizedExperiment 2021-01-27 <NA>
#12 SummarizedExperiment 2021-01-27 <NA>
#13 DFrame 2021-01-27 <NA>
#14 SummarizedExperiment 2021-01-27 <NA>
#15 SummarizedExperiment 2021-01-27 <NA>
#16 SummarizedExperiment 2023-04-24 <NA>
#17 SummarizedExperiment 2023-04-24 <NA>このコマンドを実行すると、TCGA-BRCAに関連するすべての整形済みデータセットの一覧が表示されます。
出力には、アッセイの名前、ファイルサイズ、データ形式(rdataclass)、追加日などが含まれています。
上記の出力から分かる通り、TCGA-BRCAに関連する多様なデータセットが利用可能です。
その中でも、RNASeq2GeneNorm(正規化済みRNA-Seq遺伝子発現量)と、対応する臨床情報を取り出して、生存時間解析に活用していきます。
suppressMessages({ (brca <- curatedTCGAData("BRCA", assays = c("RNASeq2GeneNorm"), dry.run = FALSE, version="2.1.1"))
})
#A MultiAssayExperiment object of 1 listed
# experiment with a user-defined name and respective class.
# Containing an ExperimentList class object of length 1:
# [1] BRCA_RNASeq2GeneNorm-20160128: SummarizedExperiment with 18300 rows and 1212 columns
#Functionality:
# experiments() - obtain the ExperimentList instance
# colData() - the primary/phenotype DataFrame
# sampleMap() - the sample coordination DataFrame
# `$`, `[`, `[[` - extract colData columns, subset, or experiment
# *Format() - convert into a long or wide DataFrame
# assays() - convert ExperimentList to a SimpleList of matrices
# exportClass() - save data to flat filesこのコードにより取得されるbrcaオブジェクトは、BioconductorのMultiAssayExperimentオブジェクトで、以下のような要素を内包しています:
experiments(brca):RNASeq2GeneNormが格納されたSummarizedExperimentオブジェクトcolData(brca): 各患者・サンプルに対応する臨床情報(生存期間、ステージ、年齢、サブタイプ情報など)sampleMap(brca): 発現データと臨床情報の対応関係(バーコードによる紐づけ)
このように、オミックスデータと臨床情報が統一されたオブジェクトとして扱えるため、面倒なマージ処理を行わずに解析を進めることができます。
データの整形と前処理
TCGAutilsパッケージを用いて、先ほどの正規化済みRNA-Seqデータと臨床情報を、解析に必要な形式へ整形します。
# 発現データを取り出す
rna <- assays(experiments(brca)[["BRCA_RNASeq2GeneNorm-20160128"]])[[1]]
# 臨床データを取り出す
clinical <- colData(brca)
# サンプルIDを12文字に統一して対応付け
colnames(rna) <- TCGAbarcode(colnames(rna))
common <- intersect(colnames(rna), rownames(clinical))
# 共通部分にフィルタ
expr_sub <- rna[, common]
clin_sub <- clinical[common, ]
# BCAS3発現量、days_to_death, vital_status を抽出
df <- data.frame( BCAS3 = as.numeric(expr_sub["BCAS3", ]), OS.time = ifelse(is.na(clin_sub$days_to_death), clin_sub$days_to_last_followup, clin_sub$days_to_death), OS.event = clin_sub$vital_status)
df <- na.omit(df)
> head(df) BCAS3 OS.time OS.event
1 8.580884 4047 0
2 9.705102 4005 0
3 8.694618 1474 0
4 9.507322 1448 0
5 9.268181 348 0
6 9.037795 1477 0ここでは、RNA-Seqデータから BCAS3(Estrogen Receptor 1) の発現量を抽出し、 生存期間(OS.time) と 生存イベント(OS.event) とともにデータフレームに整理しました。
死亡が観測されていない患者ではdays_to_last_followupを代用し、打ち切りデータも扱えるようにしています。
最後に
本記事では、GUIツールを用いず、プログラミング言語Rを用いて、TCGAの乳がんデータ(TCGA-BRCA)を取得・整形するための基本操作を体験しました。curatedTCGADataやTCGAutilsといったBioconductorパッケージを活用することで、遺伝子発現データと臨床情報を統一された形式で取得し、再現性のある解析環境を構築できることがわかりました。
さらに、MultiAssayExperimentの構造を確認し、今後の解析に必要な発現量行列や生存情報を抽出する準備が整いました。
次回は、このデータを活用して、複数の共変量と生存時間との関係をモデル化する Cox比例ハザードモデル(Cox回帰) を実行し、乳がんにおける予後に関連する要因を探索していきます。 ぜひ引き続きご覧ください。
公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!
TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます

