

ここではGROMACSのMD simulationにおける金属イオンの設定を行います。使用する力場によっては、金属イオンを設定しなければならず、その場合は自分で追加しなければいけません。Metallo protein(金属含有タンパク質)などに取り組んでいる方は参考にしてみてください!
【この記事のまとめ】
メタロプロテイン(金属含有タンパク質)の分子動力学(MD)シミュレーションに取り組む研究者を対象に、GROMACSで定義されていない金属イオン(例:Mn²⁺)のパラメータを力場に追加し、エラーなく計算を実行するための具体的な手順を解説します。
- 致命的エラーの回避:
gmx pdb2gmx実行時に発生する「Residue ‘MN’ not found」というエラーを解決するため、力場ディレクトリ(amber99sb-ildn.ff等)の内部ファイルを直接編集・カスタマイズする手法を提示。 - 物理的パラメータの厳密な設定: Lennard-Jones(LJ)ポテンシャルの$\sigma$(シグマ)や$\epsilon$(イプシロン)といった非結合パラメータについて、最新の文献(Rational Design…等)に基づいた具体的な数値の追加方法を詳説。
- トポロジーの整合性確保:
atomtypes.atpでの原子定義から、ffnonbonded.itpでの電荷設定、aminoacids.rtpでの残基登録まで、PDBファイルと力場を紐付ける一連のワークフローを公開。
この記事を読み進めることで、デフォルトの力場では扱えない金属イオンを含む系でも、原子レベルで物理学的に正しい設定を行い、高精度なシミュレーションを開始できるようになります。
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700, 64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


力場への金属イオンの追加
力場ファイルの確認
今回はamber99sb-ildn の力場に対して、Mnを追加することを行います。
例えば以下の論文で使われているHuman arginase I (HARGI)(PDB:2AEB)を参考にしてください。
QM/MM Simulations for the Broken-Symmetry Catalytic Reaction Mechanism of Human Arginase I
まずエラーが起きる箇所ですが、タンパク質の準備の時に以下のコードを行います。
gmx pdb2gmx -f protein.pdb -o protein_processed.gro -ignh
amber99sb-ildn , TIP3P を選択します。
以下のエラーが出ると思います。
-------------------------------------------------------
Program: gmx pdb2gmx, version 2024
Source file: src/gromacs/gmxpreprocess/resall.cpp (line 616)
Fatal error:
Residue 'MN' not found in residue topology database
For more information and tips for troubleshooting, please check the GROMACS
website at https://manual.gromacs.org/current/user-guide/run-time-errors.html
-------------------------------------------------------
MnがGROMACSの設定ファイルに定義されていないというエラーが出ます。
以下で解決していきます。
まずはgromacsファイルの場所を確認しましょう。以下を実行してください。
which gmx
以下のような場所が表示されると思います。
\\wsl.localhost\Ubuntu\usr\local\gromacs\share\gromacs
この階層から、さらにtop ディレクトリに移動すると、以下のように力場のファイルが見れます。

この中身の設定を変えていくわけですが、このディレクトリのものは直接変更できないので、amber99sb-ildn.ff をコピーし、作業ディレクトリに移動させ、amber99sb-ildn_Mn.ff などと名前を変更するとよいです。
Lennard-Jonesパラメータとは?
今回修正するパラメータLennard-Jones(LJ)について、説明しておきます。
数式が苦手な人は飛ばしても大丈夫です。
LJポテンシャルは、2つの非結合原子(または分子)間の相互作用エネルギーを計算する式です:
$$ V®=4\epsilon \left[ \left(\frac{\sigma}{r} \right)^{12} – \left( \frac{\sigma}{r} \right)^6 \right] $$
| 記号 | 意味 | 直感的な説明 |
|---|---|---|
| r | 原子間距離 | 2つの原子の中心間の距離 |
| ϵ\epsilonϵ | 深さ(エネルギー最小値) | 「どれだけ強く引き合うか」の強さ(単位は kJ/mol など) |
| σ\sigmaσ | 離れ具合の目安 | 原子間の「サイズ」みたいなもので、r=σr = \sigmar=σ のときにエネルギーが0になるように調整されている |
GROMACSではこのϵとσのパラメータにより、シミュレーションをしています。
金属によってはこの値が設定されていないので、これから追加していくことになる訳です。
まだまだ研究されているので、その時々に応じて、値は見直す必要がありますが、以下の論文を参考にしていることが多いので、今回も以下の論文を参考にします。
Mnイオンのトポロジーとパラメータの追加
以下のようにMnの情報を追加していきます。
-
原子タイプの定義: 力場の
atomtypes.atpファイルにマンガンイオンの原子タイプを追加します。例えば、以下のように記述します:MN 54.938ここで、
MNはマンガンの原子タイプ名、54.938は原子量です。以下のような感じです。(EditorはVS codeを使用しています。)

-
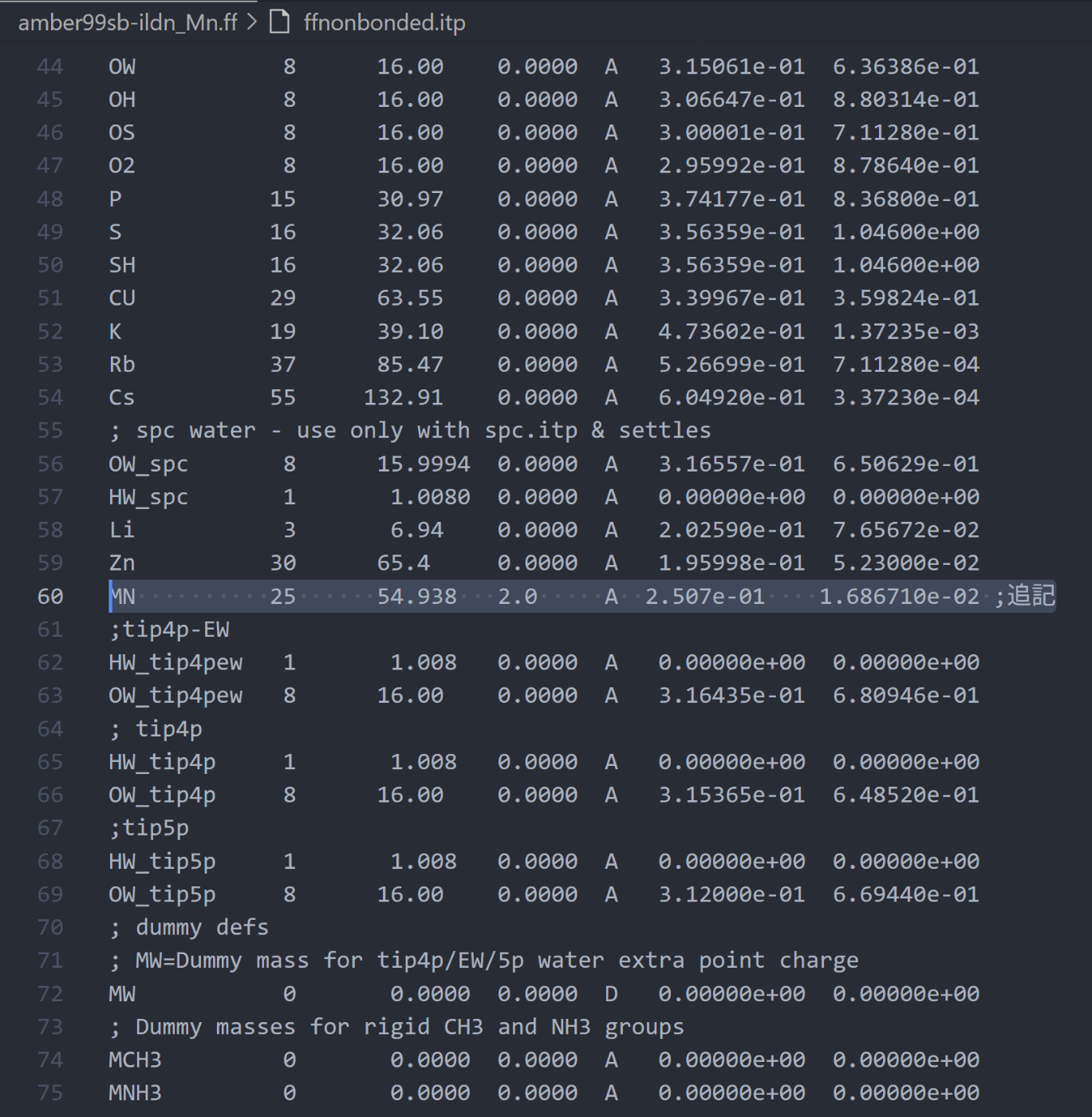
非結合パラメータの設定:
ffnonbonded.itpファイルにマンガンイオンの 12-6 Lennard-Jones(LJ)パラメータと電荷を追加します。[ atomtypes ] ; name at.num mass charge ptype sigma epsilon MN 25 54.938 2.0 A 2.507e-01 1.686710e-02 ;追記項目 内容 MN原子タイプ名。ここでは マンガンイオン(Mn²⁺) の記号として使用。 25原子番号(マンガンは 25) 54.938原子質量(amu) 2.0電荷。ここでは2価陽イオン(Mn²⁺)なので +2 Aptype。相互作用のタイプ。A = Atom 型(Lennard-Jones)0.2411σ(sigma)値。Lennard-Jonesポテンシャルの距離パラメータ(nm) 0.000650ε(epsilon)値。Lennard-Jonesポテンシャルの深さ(kJ/mol) ここで、
sigmaとepsilonの値は文献や既存の力場から取得する必要があります。今回は以下の文献を参照しています。
状況により様々な値が提唱されていますが、CMセットの値を使用すると良いと思います。
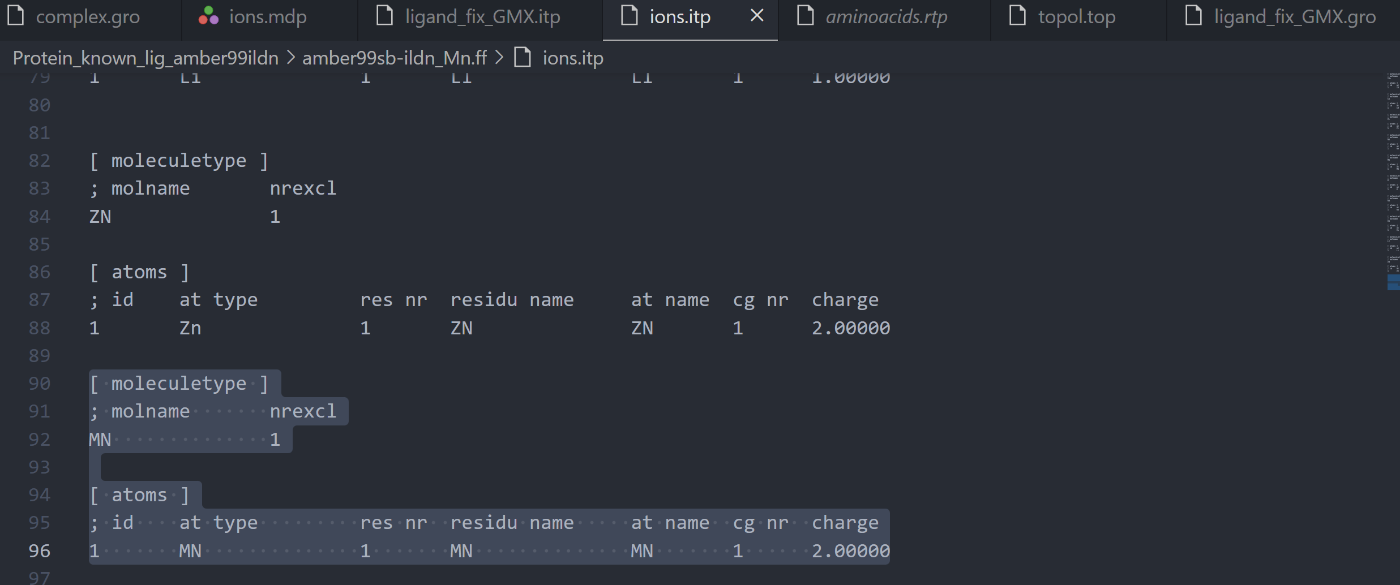
続いて`ions.itp` にマンガンのLennard-Jonesパラメータを追加してください。
```
[ moleculetype ]
; molname nrexcl
MN 1
[ atoms ]
; id at type res nr residu name at name cg nr charge
1 MN 1 MN MN 1 2.00000
```
-
残基トポロジーの追加:
aminoacids.rtpファイルにマンガンイオンの残基エントリを追加します:[ MN ] [ atoms ] MN MN 2.00000 1このエントリは、
pdb2gmxがマンガンイオンを認識するために必要です。
-
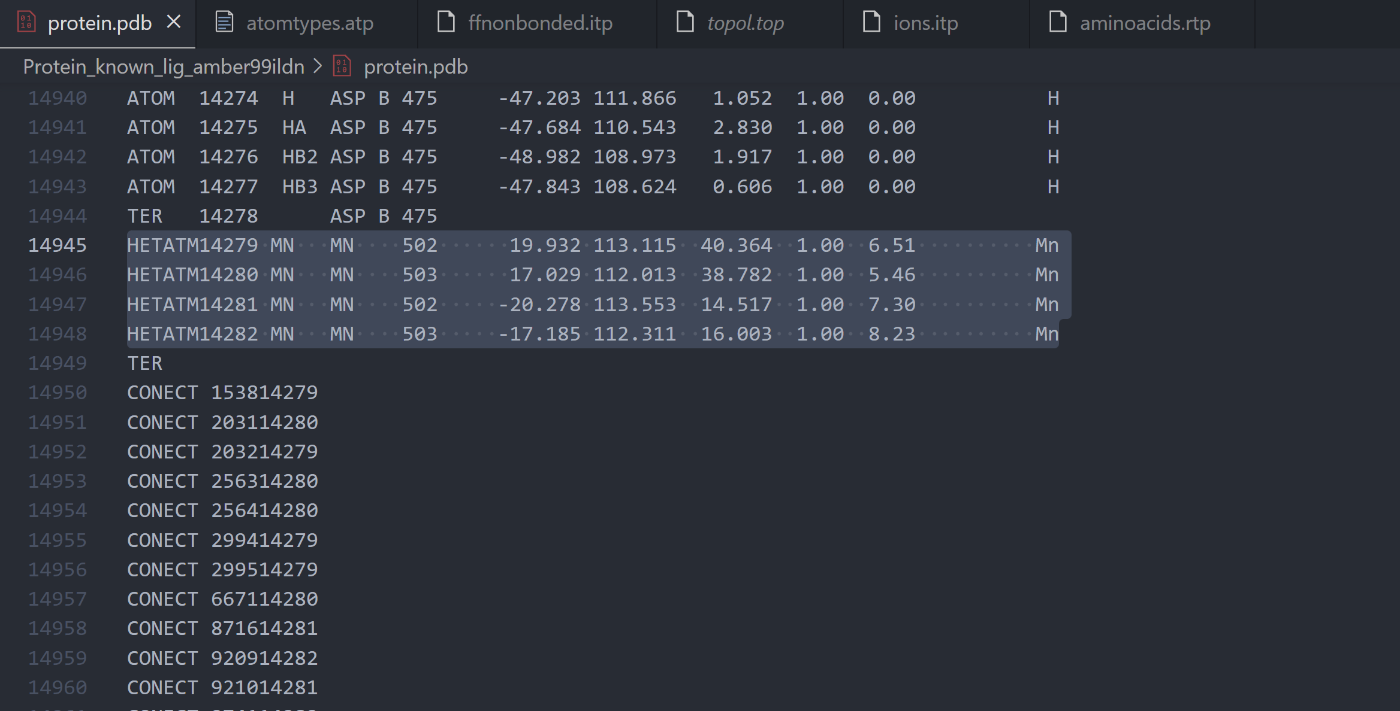
PDBファイルの修正:
- PDBファイル内のマンガンイオンの残基名が、上記で定義したもの(例:
MN)と一致していることを確認します。 - Chain IDは消す。
TERを最後に1つだけ入れる
TER 14278 ASP B 475 HETATM14279 MN MN 502 19.932 113.115 40.364 1.00 6.51 Mn HETATM14280 MN MN 503 17.029 112.013 38.782 1.00 5.46 Mn HETATM14281 MN MN 502 -20.278 113.553 14.517 1.00 7.30 Mn HETATM14282 MN MN 503 -17.185 112.311 16.003 1.00 8.23 Mn TER CONECT 153814279 CONECT 203114280 CONECT 203214279 CONECT 256314280 - PDBファイル内のマンガンイオンの残基名が、上記で定義したもの(例:
金属イオンの追加した力場でのタンパク質準備
以上を修正すると、先ほどエラー出た以下のコードが実行できます。
pdb2gmxを再実行して、新しいトポロジーファイルを生成します。
gmx pdb2gmx -f protein.pdb -o processed.gro -ignh -ff amber99sb-ildn_Mn
生成したtopol.topは以下のようになっています。other系にMnの情報が入っています。
;
; File 'topol.top' was generated
; By user: unknown (1000)
; On host: DESKTOP-5I5GHRA
; At date: Thu Jun 26 07:32:16 2025
;
; This is a standalone topology file
;
; Created by:
; :-) GROMACS - gmx pdb2gmx, 2024 (-:
;
; Executable: /usr/local/gromacs/bin/gmx
; Data prefix: /usr/local/gromacs
; Working dir: /home/shizuku/Honnma_san/Protein_Ligand/Protein_known_lig_amber99ildn
; Command line:
; gmx pdb2gmx -f protein.pdb -o processed.gro -ignh -ff amber99sb-ildn_Mn
; Force field was read from current directory or a relative path - path added.
;
; Include forcefield parameters
#include "./amber99sb-ildn_Mn.ff/forcefield.itp"
; Include chain topologies
#include "topol_Protein_chain_A.itp"
#include "topol_Protein_chain_B.itp"
#include "topol_Other.itp"
; Include water topology
#include "./amber99sb-ildn_Mn.ff/tip3p.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1
; Include topology for ions
#include "./amber99sb-ildn_Mn.ff/ions.itp"
[ system ]
; Name
protein A(仮)
[ molecules ]
; Compound #mols
Protein_chain_A 1
Protein_chain_B 1
Other 1
ここから先の処理に関しては、同じで大丈夫です!
最後に
いかがでしたでしょうか?今回はGROMACSのファイルの中身を修正していくという内容で、少し難しかったかもしれません。お疲れ様でした!金属イオンが活性に重要なメタロプロテインは自然界に多く存在するので、これを機に取り扱い方を慣れておきましょう!
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

