

この記事では、自分のパソコンで行えるMDシミュレーションに焦点を当てており、具体的にはGROMACSというソフトウェアを使用してタンパク質-低分子リガンドの動きをシミュレーションします。この記事を学ぶことで、タンパク質と低分子リガンドの動力学的な挙動や相互作用を理解することができます。ぜひ、トライしてみてください!
本記事のGROMACSのタンパク質-低分子リガンドシミュレーションは設定が長いので、前編と後編に分かれています。前編はこちらから。
【この記事のまとめ】
「GROMACS」を使用し、個人のPC環境で「タンパク質-低分子リガンド複合体」の分子動力学(MD)シミュレーションを完結させるための実践ガイド(後編)です。前編のシステム構築に続き、エネルギー最小化から平衡化、本番のシミュレーション実行、そして可視化までの具体的なパイプラインを習得できます。
- リガンドの拘束(Position Restraints)設定:
gmx make_ndxを用いたインデックスファイルの作成や、水素原子を除外した拘束条件(posre_jz4.itp)の生成手順を詳しく解説。 - トポロジーファイルの高度な編集:
topol.top内にリガンド用の拘束設定を条件付き(#ifdef)で組み込み、タンパク質とリガンドを独立して制御する手法を提示。 - シミュレーションの平衡化プロセス: 温度・圧力の安定化に向けたインデックスグループの最適化(
0 & ! a H*による原子選択など)といった、エラーを防ぐための具体的なコマンド操作を網羅。
この記事を読み進めることで、T4リゾチームと低分子(2-プロピルフェノール)を例とした動的な相互作用解析を自力で実行し、in silico創薬研究の核心となる技術を習得できます。
macOS Ventura(13.2.1), PyMOL 2.5.4., Homebrew 4.0.17, Python 3.7.3, NetworkX 2.3, Avogadro, UCSF Chimera-1.17.3-mac64

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


MD シミュレーションとは?
MDシミュレーションは、分子の動きや相互作用を計算機上で再現する手法です。分子の構造と相互作用を記述する力場(potential function)を使用し、時間の経過とともに分子の運動を予測します。これにより、物質の性質や相互作用の理解、材料設計、生体分子の研究などに応用されます。
GROMACSは、MDシミュレーションを実行するためのソフトウェアの一つです。GROMACSは高性能な計算を行うための最適化が施されており、広く使われている信頼性の高いツールです。GROMACSを使うことで、分子のダイナミクスや相互作用をシミュレーションすることができます。
この記事では実際にGROMACSを使って、自分のパソコンでタンパク質(T4リゾチームL99A/M102Q)とリガンド(2-プロピルフェノール)の複合体のMDシミュレーションをを行なっていきます。
MD シミュレーションの流れ
- タンパク質のトポロジーファイルの準備
- リガンドのトポロジーファイルの準備
- 複合体のトポロジーファイルの準備
- ボックスを定義し、溶媒を加える
- イオンを加える
- エネルギーの最小化(後編はここから)
- 平衡化
- MD シミュレーション
- 後処理
- MD シミュレーションの可視化
前編でイオンを加えるまでやったので、平衡化から行なっていきます。
続いて、平衡化を行なっていきます。
平衡化
以下のコードを実行してください。
gmx make_ndx -f jz4.gro -o index_jz4.ndxgmx make_ndx: GROMACSのツールで、インデックスファイルを生成するためのものです。インデックスファイルは、原子や分子のグループを定義するために使用されます。これにより、特定の原子や分子のグループに対して操作を簡単に実行できるようになります。f jz4.gro: このオプションとパラメータは、入力として使用する座標ファイルを指定します。この場合、**jz4.groという名前のファイルが使われます。.gro**ファイルはGROMACSの座標ファイル形式で、原子の位置情報を持っています。o index_jz4.ndx: このオプションとパラメータは、出力として生成されるインデックスファイルの名前を指定します。この場合、**index_jz4.ndx**という名前のファイルが作成されます。
コマンドを実行すると、ユーザーは対話的にインデックスグループを作成したり、既存のグループを組み合わせたりすることができます。この特定のコマンドは、**jz4.gro**ファイルに基づいてインデックスグループを生成する準備ができていることを示しています。
以下のような感じで出てくると思います。
Reading structure file
Going to read 0 old index file(s)
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups... 0 System : 22 atoms 1 Other : 22 atoms 2 JZ4 : 22 atoms nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups 'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues 't': atom type '|': or 'keep' nr 'splitat' nr 'h': help 'r': residue 'res' nr 'chain' char "name": group 'case': case sensitive 'q': save and quit 'ri': residue index
>0 & ! a H*
を打ち、Enterを押してください。
その後、qを押し、Enterを押してください。
この出力は、gmx make_ndx コマンドを使用してGROMACSのインデックスファイルを生成または編集する際の対話的セッションを示しています。以下、詳細に説明します。
Reading structure file: 指定された座標ファイル(この場合jz4.gro)を読み込んでいます。Analysing residue names: ファイル内の残基の名前を解析しています。There are: 1 Other residues: ファイル内に1つの”Other”タイプの残基が存在しています。これは、通常のプロテイン、DNA、RNA、水分子ではない残基を指します。Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...: 分類されていない残基を解析し、異なるグループに分けています。0 System, 1 Other, 2 JZ4: それぞれのグループとその中の原子の数を示しています。この場合、**SystemとJZ4とOther**の3つのグループがあり、それぞれに22の原子が含まれています。nr : group ... 'q': save and quit: これはインデックスファイルの編集時に使用できるコマンドのリストです。> 0 & ! a H: このコマンドは、グループ0 (System)の原子を選択し、その中で名前が**H*(すなわち、水素原子)であるものを除外することを意味しています。具体的には、&はAND操作を、!**はNOT操作を示します。この操作の結果、水素でない原子だけが新しいグループとして選択されます。Copied index group 0 'System': グループ0がコピーされたことを示しています。Found 12 atoms with name H: **System**グループ内に12個の水素原子が見つかったことを示しています。Complemented group: 10 atoms: 12個の水素原子を除外すると、10個の原子が残ります。Merged two groups with AND: 22 10 -> 10: オリジナルのグループ(22原子)と補完されたグループ(10原子)がAND操作で結合され、結果として10原子のグループが生成されました。> q: このコマンドで、インデックスファイルの編集を終了し、変更を保存して終了します。
総じて、このセッションは、jz4.gro ファイルから水素を除外した新しいインデックスグループを作成するプロセスを示しています。
続いて、以下のコードを打ってみてください。
gmx genrestr -f jz4.gro -n index_jz4.ndx -o posre_jz4.itp -fc 1000 1000 1000gmx genrestr: GROMACSのユーティリティの一つで、位置の拘束(position restraints)を生成するためのものです。位置の拘束は、シミュレーション中に特定の原子をその場所に「固定」するために使用されます。これは、たとえば分子の特定の部分を動かさずに、残りの部分だけを動かすシミュレーションを行いたい場合などに有用です。
コマンドのオプションについて説明します。
f jz4.gro: 入力として使用する座標ファイルを指定します。このファイルは、位置の拘束を生成したい分子の構造を含む必要があります。n index_jz4.ndx: インデックスファイルを指定します。このファイルは、前に**gmx make_ndx**コマンドを使用して作成されました。インデックスファイルは、原子の特定のグループやサブセットを定義するために使用されます。o posre_jz4.itp: 出力として生成される位置の拘束ファイルの名前を指定します。このファイルは、後でトポロジーファイル(通常**topol.top**など)にインクルードされ、シミュレーション中に位置の拘束を適用するために使用されます。fc 1000 1000 1000: これは、x, y, そしてz方向それぞれに適用する拘束の力定数を指定するものです。この場合、すべての方向での拘束の力定数は1000 kJ/mol/nm^2として指定されています。力定数が大きいほど、拘束された原子はその位置に強く固定されます。
このコマンドの実行により、**jz4.groに含まれる原子の位置を固定するための指示がposre_jz4.itp**ファイルに保存されます。
このように出力されると思います。
Reading structure file
Select group to position restrain
Group 0 ( System) has 22 elements
Group 1 ( Other) has 22 elements
Group 2 ( JZ4) has 22 elements
Group 3 ( System_&_!H*) has 10 elements
Select a group:3 を選択してください。
続いて、topol.topファイルに以下の赤字を付け加えてください。
これは二つの条件で行うことができます。
一つ目の条件はこちらです。今回はこちらの方法で行います。
もし単純にタンパク質が拘束されているときにリガンドも拘束したい場合は、指定された場所に以下の行をトポロジーに追加します:
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
; Include ligand topology
#include "jz4.itp"; Ligand position restraints
#ifdef POSRES
#include "posre_jz4.itp"
#endif; Include water topology
#include "./charmm36-jul2022.ff/tip3p.itp"二つ目の条件はこちらです。
平衡化の間にもう少し制御を持ちたい、つまりタンパク質とリガンドを独立して拘束したい場合は、異なる#ifdefブロックでリガンドの位置拘束ファイルの追加を制御できます:
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
; Include ligand topology
#include "jz4.itp"; Ligand position restraints
#ifdef POSRES_LIG
#include "posre_jz4.itp"
#endif; Include water topology
#include "./charmm36-jul2022.ff/tip3p.itp"続いて、温度を制御するために以下のコードを実行してください。
gmx make_ndx -f em.gro -o index.ndxgmx make_ndx:GROMACSのツールで、インデックスファイルを生成するコマンドです。このインデックスファイルは、特定の原子や分子のグループを指定するために使います。これにより、後続のシミュレーションや分析で特定のグループに対して操作を適用することができます。f em.gro:このオプションは、入力として用いる構造ファイル(GROファイル)を指定します。ここでは**em.gro**という名前のファイルを使用します。GROファイルはGROMACSで使われる一般的な構造ファイルのフォーマットで、システムの原子の座標やベロシティ(速度)などの情報を含むことができます。o index.ndx:このオプションで、出力されるインデックスファイルの名前を指定します。ここでは**index.ndx**という名前のファイルに結果が保存されます。
このコマンドを実行すると、ユーザーはプロンプトを介してさまざまな原子や残基のグループを定義したり、既存のグループを組み合わせたりすることができます。最終的に、指定されたインデックスグループが**index.ndx**ファイルに保存されます。
次のように出力されます。
0 System : 33506 atoms 1 Protein : 2614 atoms 2 Protein-H : 1301 atoms 3 C-alpha : 163 atoms 4 Backbone : 489 atoms 5 MainChain : 651 atoms 6 MainChain+Cb : 803 atoms 7 MainChain+H : 813 atoms 8 SideChain : 1801 atoms 9 SideChain-H : 650 atoms 10 Prot-Masses : 2614 atoms 11 non-Protein : 30892 atoms 12 Other : 22 atoms 13 JZ4 : 22 atoms 14 CL : 6 atoms 15 Water : 30864 atoms 16 SOL : 30864 atoms 17 non-Water : 2642 atoms 18 Ion : 6 atoms 19 Water_and_ions : 30870 atoms nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups 'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues 't': atom type '|': or 'keep' nr 'splitat' nr 'h': help 'r': residue 'res' nr 'chain' char "name": group 'case': case sensitive 'q': save and quit 'ri': residue index1 | 13 と打った後に、q を打ってください。
ここではまず、gmx make_ndx コマンドを使用して、em.gro という構造ファイルからインデックスグループを生成しています。インデックスグループは、後続のシミュレーションや分析において特定の原子や分子のグループを指定するために使用します。
リストの中から、以下のグループが自動的に生成されました:
0 System: システム全体の原子。合計33,506原子。1 Protein: タンパク質の原子。2,614原子。13 JZ4: JZ4という分子。22原子。- その他のグループもありますが、この解説のコンテキストにおいては、上記のグループが最も関連しています。
プロンプトでユーザーによって入力されたコマンドの解説:
> 1 | 13:1はタンパク質の原子を示すグループであり、13はJZ4の原子を示すグループです。|はOR操作を表します。この操作を使って、2つのグループを合併して新しいグループを作成します。- このコマンドにより、タンパク質とJZ4の原子が合併され、新しいインデックスグループが作成されました。この新しいグループには、2,614原子(タンパク質)と22原子(JZ4)の合計2,636原子が含まれます。
> q:- これは”quit”を意味し、操作を終了してインデックスファイルを保存します。
この操作の結果として、タンパク質とJZ4が同じインデックスグループに含まれるようになりました。これは、後のシミュレーションステップで、両方のエンティティを同時に考慮することを容易にするためです。
続いて以下のコードを実行してください。
nano nvt.mdpnano nvt.mdp というコマンドは、nvt.mdp という名前のテキストファイルを nano エディタで開くことを意味しています。
こちらのページをコピペしてください。元データはこちらから入手できます。
ctrl X→Y→Enterを押して保存してください。
続いて以下のコードを実行してください。
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tprこのコマンドは、GROMACSという分子動力学シミュレーションのソフトウェアを使って、シミュレーションを実行する前に必要な前処理を行います。具体的には、grompp (GROMACS pre-processor)というツールを使用して、シミュレーションの入力パラメータと初期構造を組み合わせて、実際のシミュレーションを実行するためのバイナリファイルを生成します。
コマンドの各オプションを詳しく見ていきましょう。
f nvt.mdp: これは使用する.mdpファイルを指定します。このファイルはシミュレーションのパラメータを含むテキストファイルです。ここでは、定温・定容(NVT)アンサンブルの条件でのシミュレーションに関する設定が含まれていると思われます。c em.gro: これは初期のコンフォメーション(構造)を含む.groファイルを指定します。この場合、**em.gro**という名前のファイルが使用されます。このファイルはエネルギー最小化された構造を持っている可能性があります。r em.gro: 参照構造を指定します。一部のシミュレーションでは、特定の原子の位置を初期位置として参照する必要があります。ここでは、エネルギー最小化された構造が再び参照として使用されています。p topol.top: これは使用するトポロジーファイルを指定します。トポロジーファイルは、システムの分子の情報や結合、角度、トルションなどの相互作用を記述しています。n index.ndx: インデックスファイルを指定します。このファイルには、特定の原子や残基のグループがリストされており、シミュレーション中にこれらのグループに特定の操作を適用することができます。o nvt.tpr: これは出力ファイルの名前を指定します。**gromppは、すべての入力情報を組み合わせて、実際のシミュレーションを実行するためのバイナリファイル.tprを生成します。この例では、出力ファイルの名前はnvt.tpr**となります。
要するに、このコマンドはシミュレーションのための入力データとパラメータを組み合わせ、実際のシミュレーションを実行するための準備を行っています。
続いて、以下のコードを打ってください。
gmx mdrun -deffnm nvtこのコマンドは、GROMACSという分子動力学シミュレーションのソフトウェアを用いて、実際のシミュレーションを実行するものです。具体的には、mdrun (molecular dynamics run)というツールを使用して、事前に**grompp**で作成された.tprファイルを基にシミュレーションを実行します。
以下は、コマンドの各オプションについての詳細な説明です。
deffnm nvt: これは、入力および出力ファイルの「デフォルトの名前」を指定するオプションです。このオプションを用いることで、入力ファイル(例:nvt.tpr)だけでなく、出力ファイル(例:nvt.log、nvt.trr、nvt.edrなど)もこの名前をベースにして生成されます。
このコマンドが実行されると、**nvt.tprという名前のファイルを基にシミュレーションが開始されます。そして、シミュレーションの進行に応じて、エネルギー、トラジェクトリ、ログファイルなどがnvt**というプレフィックスを持つ名前で出力されます。
要するに、このコマンドは、**nvt.tpr**という入力ファイルを基に、分子動力学シミュレーションを実際に実行し、結果を関連する出力ファイルに保存するものです。
ここではシミュレーションを行なっているので、しばらく時間がかかります。
続いて、次の設定を行います。
以下のコードを実行してください。
nano npt.mdpこのコマンドを実行すると、nano エディタが起動し、npt.mdp ファイルの内容が表示されます。ファイルがすでに存在していれば、その内容がエディタ上に表示されますが、存在しない場合は、新しい空のファイルが作成されます。
こちらのページをコピペしてください。元データはこちらから入手できます。
ctrl X→Y→Enterを押して保存してください。
続いて、以下のコードを実行してください。
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tprこのコマンドは、GROMACSという分子動力学シミュレーションソフトウェアの一部です。具体的には、シミュレーションを実行する前の準備段階で使用される**grompp**(GROMACS preprocessor)というツールを呼び出しています。このツールは、シミュレーションの設定、構造、トポロジーなどの情報を取り入れて、実際のシミュレーションを行うための入力ファイル(.tprファイル)を作成します。
具体的なオプションとその説明は以下の通りです:
f npt.mdp:シミュレーションのパラメータを含むMDPファイル(ここでは**npt.mdp**)を指定します。このファイルは、シミュレーションの各種設定や条件を定義します。c nvt.gro:開始構造として使用する座標ファイル(ここでは**nvt.gro**)を指定します。t nvt.cpt:シミュレーションの続きをする場合や、前のシミュレーションの結果を利用する場合に使うチェックポイントファイル(ここでは**nvt.cpt**)を指定します。r nvt.gro:リファレンス構造として使用する座標ファイル(ここでは**nvt.gro**)を指定します。このオプションは、特定のアトムの位置を初期構造としてリセットしたい場合や、位置の拘束を使用する場合に重要です。p topol.top:シミュレーションで使用するトポロジーファイル(ここでは**topol.top**)を指定します。このファイルには、分子の接続情報や力場パラメータなどが含まれます。n index.ndx:インデックスファイル(ここでは**index.ndx**)を指定します。このファイルには、アトムのグループを定義した情報が含まれます。o npt.tpr:出力として得られるバイナリのランプラグファイル(.tprファイル)の名前を指定します。このファイルは、次のステップである**mdrun**コマンドを実行する際に使用されます。
このコマンドを実行することで、**npt.tpr**という名前の.tprファイルが作成され、これをもとにGROMACSのシミュレーションが行われます。
もし警告が出て、次の操作に進めない場合は**-maxwarn 1** を足して、警告を無視するコードで実行します。以下の通りです。
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr -maxwarn 1続いて、以下のコードを実行してください。
gmx mdrun -deffnm nptこのコマンドは、GROMACSという分子動力学シミュレーションソフトウェアを用いてシミュレーションを実行する際に使われるものです。具体的には、**mdrun**というツールを呼び出しています。
コマンドの詳細について説明します:
gmx mdrun:GROMACSの主要なシミュレーションツールである**mdrun**を実行します。これにより、動力学シミュレーションやエネルギー最小化、分子動力学シミュレーションが行われます。deffnm npt:出力ファイルの名前のプレフィックスを指定します。このオプションにより、出力ファイル(トラジェクトリ、エネルギー、ログなど)のプレフィックスとして**nptが使用されます。例えば、npt.xtc、npt.log、npt.edr**などのファイルが生成されます。
具体的には、このコマンドは、**npt.tprという名前の.tprファイル(前のgrompp**コマンドの出力)を入力として受け取り、シミュレーションを実行します。このシミュレーションの結果、分子の動きやエネルギーの変動などの情報が保存されるファイルが生成されます。
このコマンドを実行することで、指定した条件とパラメータに基づいて分子動力学シミュレーションが行われ、シミュレーションの結果を含むさまざまな出力ファイルが得られます。
このステップでは4時間ほど待ちます。
MD シミュレーション
ここまで様々な設定お疲れ様でした。いよいよMDシミュレーションを使っていきます。
続いて以下のコードを実行してください。
nano md.mdpこのコマンドを実行すると、nano エディタが起動し、md.mdpファイルの内容が表示されます。ファイルがすでに存在していれば、その内容がエディタ上に表示されますが、存在しない場合は、新しい空のファイルが作成されます。
こちらのページをコピペしてください。元データはこちらから入手できます。
ctrl X→Y→Enterを押して保存してください。
続いて、以下のコードを実行してください。
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_10.tprこのコマンドは、GROMACSの分子動力学シミュレーションソフトウェアの一部として使われるものです。**gromppツールは、シミュレーションを実行する前の準備ステップで使用されます。具体的には、入力ファイルを読み込み、シミュレーションに必要なすべての情報を持つ実行可能な.tpr**ファイルを作成します。
コマンドの各部分について詳しく説明します:
gmx grompp:GROMACSの**grompp**ツールを呼び出します。これは、シミュレーションの準備ツールで、いくつかの入力ファイルを組み合わせて、実際のシミュレーションを実行するための.tprファイルを生成します。f md.mdp:シミュレーションのパラメータを指定する.mdpファイルの名前を指定します。c npt.gro:初期構造を示す.groファイルの名前を指定します。t npt.cpt:前のシミュレーションの状態を示す.cptファイルの名前を指定します。このファイルは、シミュレーションの途中から再開したい場合や、シミュレーションの連続的な実行をする場合に使用されます。p topol.top:システムのトポロジーを示す.topファイルの名前を指定します。n index.ndx:さまざまな分子グループを示す.ndxファイルの名前を指定します。o md_0_10.tpr:出力として生成される.tprファイルの名前を指定します。この.tprファイルは、次に**mdrun**ツールを使用してシミュレーションを実行する際に使用されます。
このコマンドを実行することで、指定された入力ファイルを元に、シミュレーションを実行するための**md_0_10.tpr**という名前の.tprファイルが生成されます。
gmx mdrun -deffnm md_0_10このコマンドは、GROMACSの分子動力学シミュレーションソフトウェアの一部として使用されます。**mdrun**ツールは、実際に分子動力学シミュレーションを実行するためのものです。
コマンドの詳細を以下に説明します:
gmx mdrun:GROMACSの**mdrun**ツールを呼び出します。これは、分子動力学シミュレーションを実行する主要なツールです。deffnm md_0_10:シミュレーションに関連する入力および出力ファイルのデフォルト名を指定します。このオプションが指定されると、指定された名前(ここでは**md_0_10)を持つファイルが使用されるか、または生成されます。具体的には、md_0_10.tpr**という名前の.tprファイルが入力として読み込まれ、シミュレーションが実行されます。
シミュレーションが完了すると、以下のような一連のファイルが生成される可能性があります:
md_0_10.trr:軌道ファイル。これは、シミュレーション中のすべてのフレームの原子の位置、速度、および力を含む可能性があります。md_0_10.xtc:圧縮された軌道ファイル。これは、シミュレーション中の原子の位置のみを含む可能性があります。md_0_10.edr:エネルギーファイル。これは、シミュレーション中の各ステップのエネルギーを記録するファイルです。md_0_10.log:ログファイル。これは、シミュレーションの進行に関する情報や警告を記録するためのファイルです。md_0_10.gro:シミュレーションの最後のフレームの構造を示す.groファイル。
このコマンドを実行することで、**md_0_10.tpr**ファイルに基づいて分子動力学シミュレーションが実行され、関連する出力ファイルが生成されます。
これにて、MDシミュレーションは終了です。めちゃくちゃ時間かかるので、覚悟が必要です。私は4日くらいかかりました。
以下のメッセージが出るまで待ちましょう!
Dynamic load balancing report: DLB got disabled because it was unsuitable to use. Average load imbalance: 4.0%. The balanceable part of the MD step is 63%, load imbalance is computed from this. Part of the total run time spent waiting due to load imbalance: 2.5%.
WARNING: A total of 0.000000 CPU cycles was recorded, so mdrun cannot print a time accounting Core t (s) Wall t (s) (%) Time: 2598405.385 259840.540 1000.0 3d00h10:40 (ns/day) (hour/ns)
Performance: 3.325 7.218
GROMACS reminds you: "Culture eats strategy for breakfast" (Peter Drucker)後処理
MDシミュレーションが終わると、現在のディレクトリにmd_0_10.拡張子 のファイルがいくつも生成していると思います。これでもシミュレーションの解析はできるのですが、分子が「壊れた」ように見えたり、ボックスを跨いで前後に「ジャンプ」することがあります。以下のコードで直します。
次に以下のコードを実行してください。
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compacts md_0_10.tpr: シミュレーションの設定情報が含まれている**.tpr**ファイルを指定します。このファイルは、シミュレーションの「レシピ」のようなものです。f md_0_10.xtc: 入力として使用するトラジェクトリファイル(.xtc)を指定します。このファイルには、シミュレーション中の各時間ステップでの分子の座標が含まれています。o md_0_10_center.xtc: 出力トラジェクトリファイル(.xtc)の名前を指定します。このファイルには、変換後の座標が保存されます。center: このオプションは、タンパク質(または他の分子)をユニットセルの中心に配置するために使用されます。pbc mol: このオプションは、周期的境界条件(PBC)を考慮して分子をユニットセル内に「ラップ」します。これにより、分子がユニットセルの境界を越えて「ジャンプ」するのを防ぎます。ur compact: このオプションは、出力されるユニットセルの形状を「コンパクト」(通常は菱形十二面体)にするために使用されます。
このコマンドは、特に長いシミュレーションで分子がユニットセルを跨いで「ジャンプ」する場合や、分子が「壊れて」見える場合に非常に便利です。それによって、分子の座標がより解釈しやすく、分析しやすくなります。
次のように出力されると思います。
elect group for centering
Group 0 ( System) has 33506 elements
Group 1 ( Protein) has 2614 elements
Group 2 ( Protein-H) has 1301 elements
Group 3 ( C-alpha) has 163 elements
Group 4 ( Backbone) has 489 elements
Group 5 ( MainChain) has 651 elements
Group 6 ( MainChain+Cb) has 803 elements
Group 7 ( MainChain+H) has 813 elements
Group 8 ( SideChain) has 1801 elements
Group 9 ( SideChain-H) has 650 elements
Group 10 ( Prot-Masses) has 2614 elements
Group 11 ( non-Protein) has 30892 elements
Group 12 ( Other) has 22 elements
Group 13 ( JZ4) has 22 elements
Group 14 ( CL) has 6 elements
Group 15 ( Water) has 30864 elements
Group 16 ( SOL) has 30864 elements
Group 17 ( non-Water) has 2642 elements
Group 18 ( Ion) has 6 elements
Group 19 ( Water_and_ions) has 30870 elements
Select a group:中心にはタンパク質を置きたいので、”1”を選択してください。
続いて、
Select group for output
Group 0 ( System) has 33506 elements
Group 1 ( Protein) has 2614 elements
Group 2 ( Protein-H) has 1301 elements
Group 3 ( C-alpha) has 163 elements
Group 4 ( Backbone) has 489 elements
Group 5 ( MainChain) has 651 elements
Group 6 ( MainChain+Cb) has 803 elements
Group 7 ( MainChain+H) has 813 elements
Group 8 ( SideChain) has 1801 elements
Group 9 ( SideChain-H) has 650 elements
Group 10 ( Prot-Masses) has 2614 elements
Group 11 ( non-Protein) has 30892 elements
Group 12 ( Other) has 22 elements
Group 13 ( JZ4) has 22 elements
Group 14 ( CL) has 6 elements
Group 15 ( Water) has 30864 elements
Group 16 ( SOL) has 30864 elements
Group 17 ( non-Water) has 2642 elements
Group 18 ( Ion) has 6 elements
Group 19 ( Water_and_ions) has 30870 elements
Select a group:アウトプットにはSystemの”0”を選択してください。
続いて、
gmx trjconv -s md_0_10.tpr -f md_0_10_center.xtc -o md_0_10_fit.xtc -fit rot+transこの**gmx trjconv**コマンドは、GROMACSの分子動力学シミュレーションのトラジェクトリ(分子の時間に対する位置)に対して回転と並進のフィッティングを行うために使用されます。以下に各オプションの説明をします。
s md_0_10.tpr: このオプションで、シミュレーションの設定情報が含まれている**.tpr**ファイルを指定します。このファイルは、シミュレーションの「レシピ」のようなものです。f md_0_10_center.xtc: このオプションで、入力として使用するトラジェクトリファイル(.xtc)を指定します。このファイルには、シミュレーション中の各時間ステップでの分子の座標が含まれています。o md_0_10_fit.xtc: このオプションで、出力トラジェクトリファイル(.xtc)の名前を指定します。このファイルには、フィッティング後の座標が保存されます。fit rot+trans: このオプションで、回転(rot)と並進(trans)のフィッティングを行うことを指定します。これにより、タンパク質(または他の分子)の全体的な回転と並進が除去され、その結果、分子の相対的な動きだけがトラジェクトリに残ります。
このコマンドは、特に分子の動きを視覚化する際や、分子間の相対的な動きを分析する際に有用です。回転と並進のフィッティングを行うことで、分子の動きがより明確になり、その結果、分析が容易になります。
以下のように出力されてきます。
Select group for least squares fit
Group 0 ( System) has 33506 elements
Group 1 ( Protein) has 2614 elements
Group 2 ( Protein-H) has 1301 elements
Group 3 ( C-alpha) has 163 elements
Group 4 ( Backbone) has 489 elements
Group 5 ( MainChain) has 651 elements
Group 6 ( MainChain+Cb) has 803 elements
Group 7 ( MainChain+H) has 813 elements
Group 8 ( SideChain) has 1801 elements
Group 9 ( SideChain-H) has 650 elements
Group 10 ( Prot-Masses) has 2614 elements
Group 11 ( non-Protein) has 30892 elements
Group 12 ( Other) has 22 elements
Group 13 ( JZ4) has 22 elements
Group 14 ( CL) has 6 elements
Group 15 ( Water) has 30864 elements
Group 16 ( SOL) has 30864 elements
Group 17 ( non-Water) has 2642 elements
Group 18 ( Ion) has 6 elements
Group 19 ( Water_and_ions) has 30870 elements
Select a group:ここではタンパク質の主鎖に対してフィッティングを行いたいので、4を選択します。続いて出てくるoutputにはSystemを意味する”0”を選択してください。
MD シミュレーションの可視化
それではこのMDシミュレーションを可視化してみましょう!
ソフトウェアとしてUCSF Chimeraを使います。こちらからダウンロードしてください。
UCSF Chimeraは、タンパク質や核酸などの生物分子の3D構造を視覚化、分析するためのソフトウェアです。多くのファイル形式をサポートし、分子間の相互作用や距離を計算できます。教育、研究、薬剤設計に広く使用されています。
Tools→MD/Ensemble Analysis→MD Movieを選んでください。
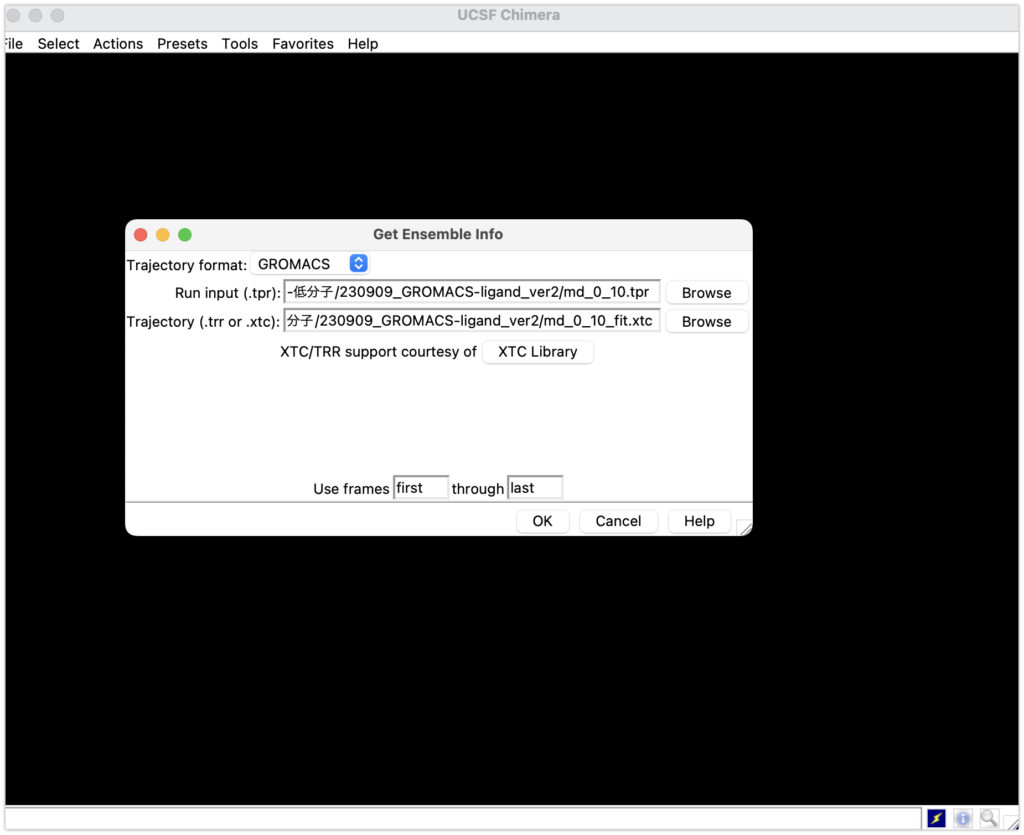
続いて、Run inputにmd_0_10.tpr 、Trajectoryにmd_0_10_fit.xtcを入れてOKを入れてください。
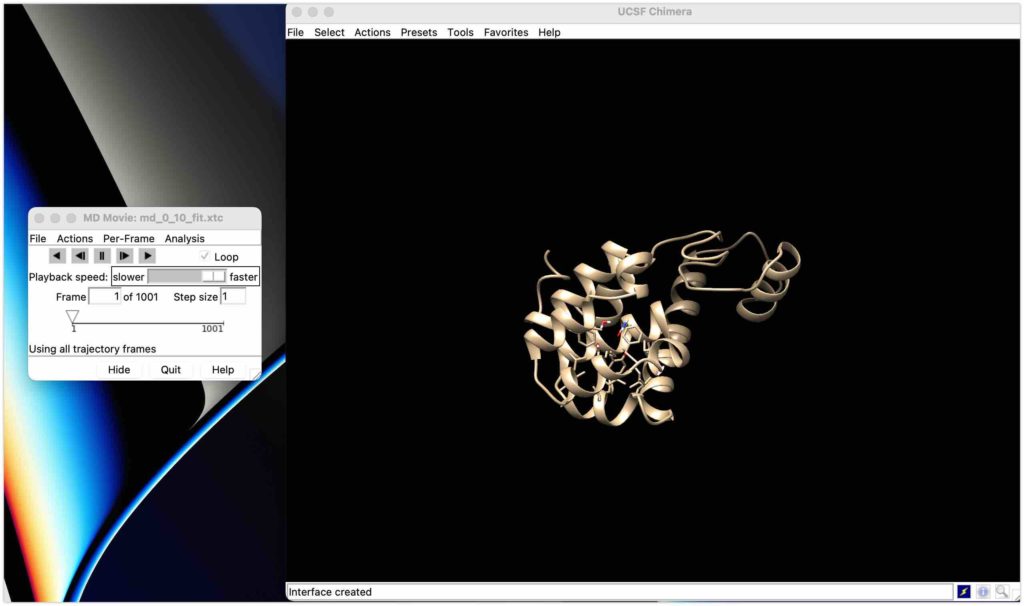
タンパク質とリガンドが出てきました!
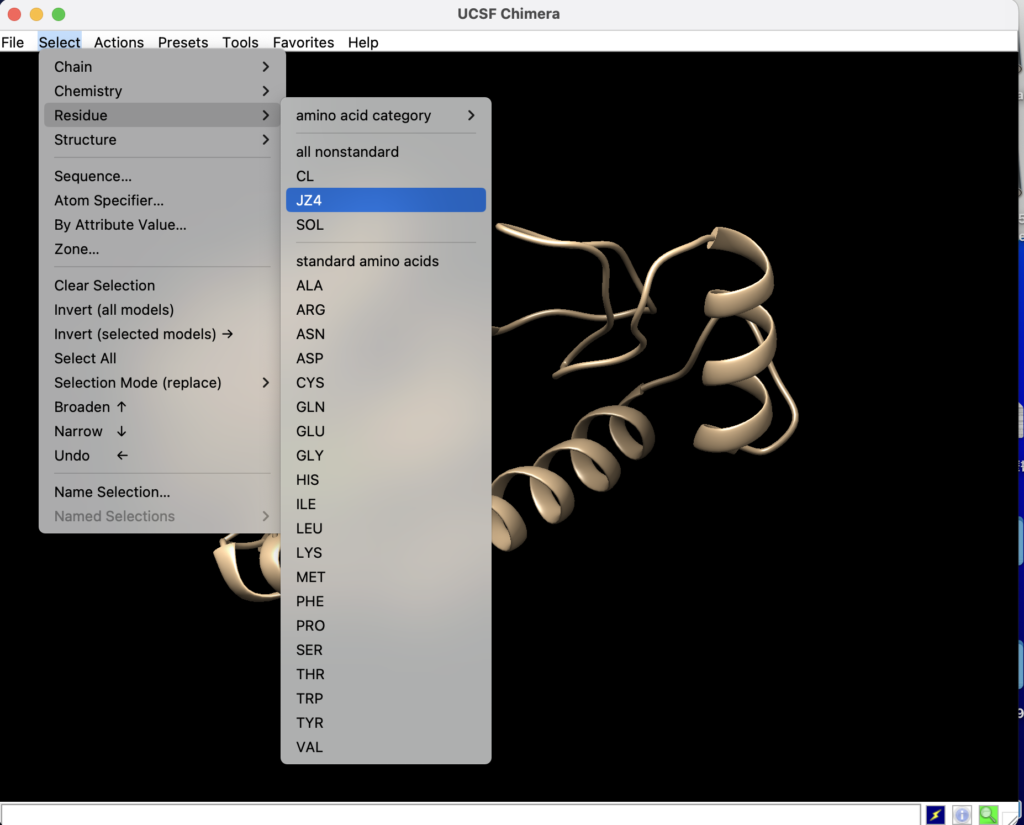
見やすくするためにリガンドをマークしましょう!
上のタブのselect→Residue→JZ4からリガンドを選びます。
最後にMD Movieのスタートボタンを押せばシミュレーションできます!
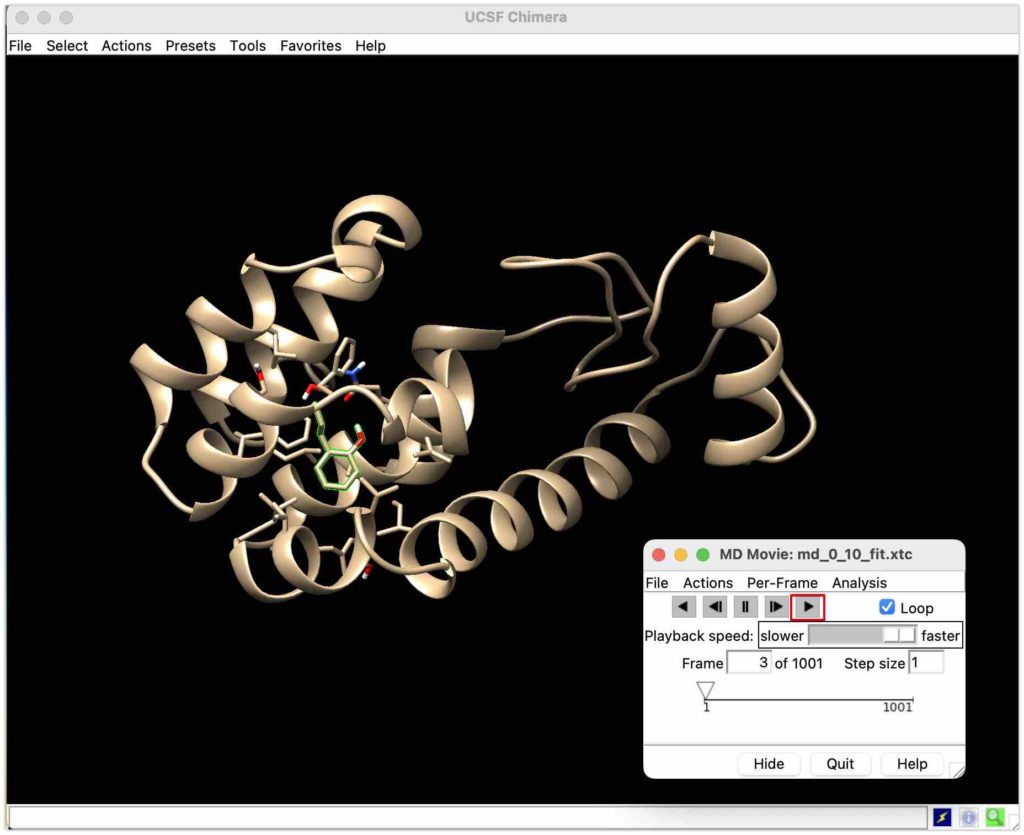
おお、すごく綺麗にシミュレーションできています!お疲れ様でした!
最後に
いかがでしたでしょうか。タンパク質とリガンドのMDシミュレーションは、タンパク質とリガンドの相互作用を明らかにし、薬物効果の理解を深めます。その結果は医薬品開発やタンパク質工学に重要な示唆を与え、新たな治療法やバイオテクノロジーの進歩につながる可能性があります。皆さんも是非試してみて、分子動力学の魅力を体験し、分子レベルの世界に触れる興奮を感じてみましょう!
参考資料
Molecular Dynamics Tutorial | Protein-Ligand Complex: GROMACS – PART 1
Molecular Dynamics Tutorial | Protein-Ligand Complex: GROMACS – PART 2
EP 11 | Post MD Simulation movie for visualization Protein-Ligand complex trajectories | Gromacs
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

