

今回は環状ペプチドの立体構造や配列のデザインから、その構造の評価まで一貫してできるBolzgenについて解説をしていきます。Bolzgenの概要・インストール方法・使い方を合わせて説明します。
【この記事のまとめ】
本記事は、創薬研究やバイオインフォマティクスに携わる方へ向けて、生成AI(潜在拡散モデル)を活用して標的タンパク質に結合する環状ペプチドをゼロから設計する革新的手法「BOLZGEN」 の仕組みと有用性を解説しています。
- 潜在拡散モデル(LDM)とRosettaの融合: 幾何学的な拡散モデルを用いてペプチドのバックボーンを生成し、物理ベースのRosettaで配列設計とエネルギー最適化を行うことで、高精度な設計を実現しています。
- 難関ターゲットでの実証: MDM2やIL-17といった重要な創薬標的に対し、既存手法を上回る設計成功率と、実験的に裏付けられた高い結合親和性(K_D値)を示しています。
- 高い安定性と多様な設計: 生成される環状ペプチドは熱安定性に優れ、多様な構造サンプリングが可能なため、従来の設計手法では到達できなかった新しい阻害剤の探索が可能です。
この記事を読むことで、AIがどのように環状ペプチドの複雑な構造を学習し、実用レベルの創薬候補へと昇華させるかの具体的なプロセスを把握できます。
Windows 11, WSL2(Ubuntu 20.04),bash
PCスペック
CPUメモリ: 16GB
GPU0: NVIDIA GeForce RTX 2070 SUPER(専用メモリ 8GB)
GPU1: Intel UHD Graphics 630(内蔵 GPU)
GPUドライバー: 581.29-desktop-win10-win11-64bit-international-dch-whql

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


Bolzgenとは?
Bolzgenとは、タンパク質の立体構造のデザインから、配列のデザイン、その親和性の予測といった構造の評価までを一貫して行えるde novoタンパク質デザインツールのことです。様々なタイプの立体構造のデザインに対応しており、環状ペプチドのデザインにも利用することができます。
同じく拡散モデルを利用する環状ペプチドデザインツールとしてはRFPeptideが挙げられますが、BolzgenはRFPeptideと比較して、一つのツールでより多くのステップを一気通貫で完結させられます。具体的には、RFPeptideの場合では立体構造のデザインからその構造の評価までを行うために、立体構造のデザインにRFPeptide、配列予測にProteinMPNN、構造の評価にAlphaHoldというように3つのツールを使う必要がありましたが、BolzgenではこれらのステップをBolzgenのみで行うことが可能です。
また、RFPeptideでは、立体構造のデザインとアミノ酸配列の構築という2つのステップが段階的に行われる為に、立体構造のデザインの際にアミノ酸配列では物理的に構築不可能な構造がデザインされてしまう可能性がありましたが、今回紹介するBolzgenでは立体構造のデザインと配列予測が統合的に行われるため、アミノ酸で構成可能な立体構造のみをデザインできる様になっています。
Bolzgenの原理
以下でツールの原理をもう少し詳しく説明します。(以下は、専門的なので読み飛ばしても大丈夫です。)
Bolzgenは、all-atom diffusion modelと呼ばれる拡散モデルを利用しています。このモデルによって、Bolzgenは構造デザインとアミノ酸配列の構築を同時に行うことができます。このモデルを簡単に説明すると、アミノ酸の形やその特徴(疎水性残基は内部に配置されやすいや、極性残基は表面に出やすいなど)を理解できるようになった立体構造生成モデル(diffusion model)です。
diffusion modelとは、特定の条件(例:たんぱく質Aに結合する)を満たす立体構造を生成するためのモデルでRFPeptideにも利用されていますが、RFPeptideのモデルは主に「骨格構造(backbone)」のみを対象としていました。つまり、主鎖原子の構造を生成することはできても、側鎖を含めた原子レベルでの精密な構造までは同時に扱えないという制約があり、この制約から上述のように生成された構造と残基の整合性が必ずしも保証されていませんでした。
Bolzgenのall-atom diffusion modelでは、残基タイプの幾何学的エンコーディングという方法を利用して、この制約を突破しています。残基タイプの幾何学的エンコーディングとは、アミノ酸の種類ごとの①形状と分子組成、②立体構造の自由度、③残基の化学的な性質(電荷や水素結合のしやすさなど)、④タンパク質立体構造下での特徴(内部に埋もれやすいか、表面に出やすか)などの特徴を機械学習モデルが扱える形に変換することです。
all-atom diffusion modelはこれを行うことにより、立体構造を生成する機械学習モデルでありながら、アミノ酸の形と特徴を理解できるようになっており、従来のモデルの様に、単に主鎖原子の座標情報だけを扱うのではなく、「その原子がどのアミノ酸残基に属しているのか、そしてそのアミノ酸はどのような特徴をもっているのか」という化学的な情報を、幾何学的な特徴として同時にモデルへ組み込むことが出来るのです。
この特徴によりBolzgenでは、構造デザインの段階で既に「この配列で折りたたみ可能か」「結合界面が安定か」といった情報を考慮した生成が行われます。結果として、従来のように構造生成後に別ツールで配列を当てはめ、さらに別ツールで評価するといった段階的プロセスに比べて、より整合性の取れたデザインが可能になります。
またBolzgenは、単なる構造生成にとどまらず、生成した候補に対して結合親和性や安定性をスコアリングする仕組みも組み込まれているため、設計から評価までを一つのフレームワークで完結させることができます。
このようにBolzgenは、環状ペプチドを含むde novoタンパク質デザインにおいて、従来よりも統合的かつ実用的なワークフローを提供するツールとして注目されています。
Bolzgenの環境構築
では早速、Bolzgenを利用して見ましょう。
まずはこちらを参考に環境構築から行っていきます。今回もWindowsのWSLを利用していきます。WSLの利用方法はこちらの記事に記載してあるので、そちらを参考にしてください。
今回のツールはGPUを利用するツールのため、まずはこちらのサイトでお使いのPCに搭載されているGPUに 対応したドライバ(Windows11用。Linux用ではありません)をDLして、Windows 上でインストーラを実行し、再起動して有効化してください。また、どのドライバーが良いのか分からない場合は、こちらのNDIVIA appをインストールすると最適なドライバーをインストールしてくれるようです(既にインストール済みの場合は不要です。)
次に、WSLのターミナルで以下のコードを実行して、環境構築をしていきます。
ターミナルを開き、以下を実行してください。途中で3回ほどEnterキーの入力を求められますので、入力することを忘れないようにしてください。
# miniconbaがインストールしていない場合は、以下を実効
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash ~/Miniconda3-latest-Linux-x86_64.sh
# 途中で3回ほどEnterキーの入力を求められますので、入力してインストールを続行してください
# 以下で初期化し、minicondaを有効にします
source ~/.bashrc
# ここまでminicondaのインストール
# condaでbgという環境を作り、pythonをインストール
conda create -n bg python=3.12 -y
conda activate bg
# boltzgenをインストール
pip install boltzgenBolzgenの実行
これで準備が整いました。Bolzgenを利用して行きます。
まずは、作業したいフォルダを決めます(私はWindowsのdocuments下に作ったbolzgenというフォルダで作業します)。
次に、Bolzgenに渡すターゲットタンパクの立体構造のPDBファイルを用意します。利用する立体構造はご興味に合わせてどのようなものでも良いですが、私はRFPeptideの解説の際に利用したタンパクファイル(7zkr_GABARAP.pdb)をダウンロードして、ターゲットタンパクとしました。
(既に目的のタンパク構造ファイルがある方も同様に、ここで立体構造ファイルをコピペで作業フォルダ内に置いておきましょう)。
7zkr_GABARAP.pdbのファイルはこちらからダウンロードできます。ダウンロードしたファイル(rfd_macro.tar.gz)のexamples>input_pdbs内にあるので、こちらのPDBファイルを作業フォルダ内に移動しておきましょう。
そして、以下のコマンドで作業したいフォルダに移動します
# 作業フォルダへ移動
cd /mnt/c/users/(あなたのWondowsのユーザー名)/documents/bolzgen/documents/RFPeptideは、ご自身が作業したいフォルダの名前に合わせて変更して下さい。そして、作りたい環状ペプチドの構造を指定するためのYAML(ヤムル)ファイルを準備します。
まずは、WSLで以下を実行して下さい。nanoというツールを起動して、こちらでYAMLファイルを作成します。
nano cyclic_cs_binding.yamlそして、開いたwindowに以下をコピペ。
entities: - file: path: /mnt/c/users/(あなたのWondowsのユーザー名)/Documents/bolzgen/7zkr_GABARAP.pdb include: - chain: { id: A } - protein: id: CP sequence: 10..20 cyclic: true
binding_types: - chain: id: A binding: 163..181その後、キーボード入力で Ctrl + O でファイルを保存。その際、ファイル名の確認を求められるので、問題なければEnterで決定し、Ctrl + X でnanoを終了してください。
このYAMLファイルはいわば環状ペプチドの設計仕様書のようなもので、どのように環状にするかや、結合したいタンパク質やその結合部位を指定することができます。
あとは以下のコマンドでbolzgenを実行します。実行の終了まで時間がかかりますが、問題がなければこれで環状ペプチドのデザインが出力されているはずです。
boltzgen run cyclic_cs_binding.yaml \ --protocol peptide-anything \ --output ./output_run1 \ --num_designs 5 \ --budget 2実行結果
作業フォルダにoutput_run1というフォルダが出来、中身が以下の様になっていれば成功です。
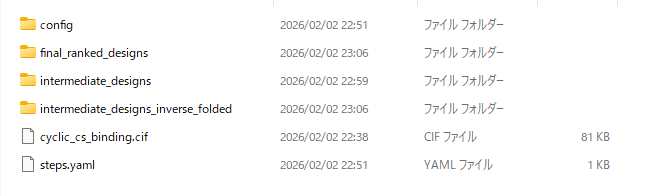
configフォルダには、今回のデザイン設定の詳細が、final_ranked_designsフォルダには、最終的にbolzgenで選ばれたデザインが、intermediate_designsフォルダには、立体構造と配列の決定の途中経過の詳細が、intermediate_designs_inverse_foldedフォルダには、配列が決定した後の途中経過の詳細が入っています。cyclic_cs_binding.cifは生成前のスタート時点の構造が、steps.yamlには今回の実行でどのような計算を行ったのかの説明が記されています。
final_ranked_designsフォルダの中身は以下の様になっています。
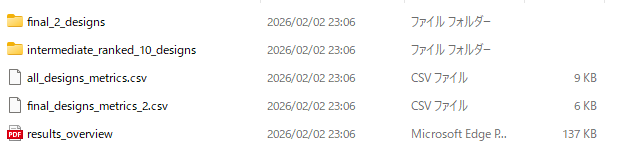
final_2_designsフォルダには、最終的に選ばれた2つの環状ペプチドデザインが、intermediate_ranked_10_designsフォルダには、上位10位までのデザインが、2つのCSVファイルにはそれぞれのデザインのスコアが、pdfファイルには、スコアやフィルタリング基準がグラフ化された今回の結果のレポートが出力されています。
2つのCSVファイルのうち、final_designs_metrics_2.csvの中には、以下の様に上位2つのデザインのスコアが入っています(CSVファイルはエクセルなどで開くと表形式で見やすく表示できます)。
実際のCSVには100種類を超える様々なスコアが並んでいますが、特に重要なスコアは以下の4つのようです(以下では、実際のCSVファイルの中身を省略して、一部を示しています。)。
| id | design_to_target_iptm | min_design_to_target_pae | filter_rmsd | liability_violations_summary |
|---|---|---|---|---|
| cyclic_cs_binding_0 | 0.72804 | 1.84653 | 1.04226 | AspCleave(pos1,sev10); ProtTryp(pos10,sev10) |
| cyclic_cs_binding_1 | 0.72693 | 1.95509 | 1.06399 | AspCleavex2(pos1-9,sev10) |
- design_to_target_iptm 設計したペプチドがターゲット(GABARAP)にどれくらい強く結合しそうか。一般に0.7以上はまずまず。0.8以上だと良い。
- min_design_to_target_pae 結合位置の誤差(不確かさ)です。 低いほどよく、一般に2以下なら良い。5以上は信頼性が低い。
- filter_rmsd 立体的に無理がないか(衝突や崩れ)を見る指標です。低いほどよく、一般に2以下なら良い。5以上は信頼性が低い。
- liability_violations_summary 分解されやすい構造があれば記載される。今回は
AspCleave(pos1,sev10); ProtTryp(pos10,sev10)などの記載があり、ASP部位やトリプシン切断部位があるとのこと。
モデルの性質上、完全に同じ結果とはなっていないはずですが、皆様はどの程度のスコアとなっていたでしょうか。
最後に、Mol* Viewerで先ほどのfinal_2_designsフォルダの中からrank1_cyclic_cs_binding_0.cifの構造を視覚的に確認してみましょう!
上記のリンクからビューワーを開き、Open fileからrank1_cyclic_cs_binding_0.cifをアップロードして、Applyをしてください。アップロードしたcifファイルが開き、bolzgenで作成した環状ペプチドと目的タンパク質の構造が確認できます。(以下の画像を参考に進めてください。)
ファイルが開くと、以下の様になっていました。生成された環状ペプチド(下の赤枠で囲った部分)が目的タンパク質の指定した箇所に結合していそうなことが確認できました。

コードの解説
上に書いたソースコードの解説をしていきます。 以下は、BoltzGen を動かすためのPython環境を作ることを目的としたコードです。
# miniconbaがインストールしていない場合は、以下を実効
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash ~/Miniconda3-latest-Linux-x86_64.sh
# 途中で3回ほどEnterキーの入力を求められますので、入力してインストールを続行してください
# 以下で初期化し、minicondaを有効にします
source ~/.bashrc
# ここまでminicondaのインストールここまでは、minicondaというツールをインストールするためのコマンドです。minicondaを利用して、環境構築をすることで簡単にPython環境を用意することが出来ます。すでにインストールしている場合は、不要です。
# condaでbgという環境を作り、pythonをインストール
conda create -n bg python=3.12 -y
conda activate bg
# boltzgenをインストール
pip install boltzgenconda create -n bg python=3.12 -y これは、bgという名前の環境をconda createで作成し、Python3.12をインストールするというコマンドです。Bolzgenはpython3.12に対応しているので、このバージョンのpythonをインストールします。-yというオプションでインストール時の確認を省略しています。
conda activate bg で、作成したbg環境を有効化しています。
pip install boltzgen で、先ほどの環境内にbolzgenをインストールしています。
以下は、ymlファイルの解説です。
entities: - file: path: /mnt/c/users/(あなたのWondowsのユーザー名)/Documents/bolzgen/7zkr_GABARAP.pdb include: - chain: { id: A } - protein: id: CP sequence: 10..20 cyclic: true
binding_types: - chain: id: A binding: 163..181まず、以下のパートは結合させるタンパク質を指定しています。path: /mnt/c/users/(あなたのWondowsのユーザー名)/Documents/bolzgen/7zkr_GABARAP.pdbでタンパク質の立体構造ファイルを指定し、- chain: { id: A } でタンパク質の鎖を指定しています。今回のタンパク質ファイル内にはA鎖のみですが、複数の鎖があった場合にはここで指定が可能です。
- protein: これは新しく作成するペプチドに関する条件を指定します。id:でペプチドの名前を指定し、sequence:で長さを指定できます。sequence: 10..20は10~20残基の長さを指定しています。cyclic: trueで環状ペプチド(C-N接続)を指定しています。
binding_types: では、結合させたい箇所を指定できます。id: AでA鎖の、binding: 163..181で163~181残基に結合するように指定しています。
ちなみに今回は、N末端とC末端をつなげて「環状ペプチド」にするように指定しましたが、以下のyamlの様に書くことで、Cys–Cys disulfide結合による環状ペプチドをしてすることが出来ます。
entities: - file: path: /mnt/c/users/(あなたのWondowsのユーザー名)/Documents/bolzgen/7zkr_GABARAP.pdb include: - chain: { id: A } - protein: id: C sequence: 1..5C6C1..5
constraints: - bond: atom1: [C, 2, SG] atom2: [C, 9, SG]
binding_types: - chain: id: A binding: 163..181この場合は、cyclic: true を使わずに、constraints: の-bond:で結合させたいCys残基を指定して下さい。また、この時に、- protein: のsequence:でC(Cys)が配列のどこかに二か所に必ず含まれるように指定しておくことを忘れないでください。
また、今回は利用しませんでしたが、以下のstructure_groups:オプションで結合させたいタンパク質の一部が可変する場合の設定も可能なようです。
structure_groups: - group: id: A visibility: 1 - group: id: A visibility: 0 res_index: 32..42最初にA鎖全体をvisibility: 1で可変構造にしないとしてした後に、再度 - group: して、res_index: でA鎖の一部(今回は32~42)を visibility: 0 で可変可能な構造として指定できます。結合で形が変わる場所や、もともと柔軟に動く場所があるタンパク質の場合はこちらのオプションを使うこともよいかもしれません。
以下は、bolzgenの実行コードです。
boltzgen run cyclic_cs_binding.yaml \ --protocol peptide-anything \ --output ./output_run1 \ --num_designs 5 \ --budget 2これは、boltzgenの実行コマンドです。
boltzgen→ BoltzGen の実行コマンド(本体)run→ 設計を開始する命令cyclic_cs_binding.yaml→ 設計のルールを書いた設定ファイルを指定-protocol→ どんな設計モードで動かすか指定するオプション。今回は、peptide-anything
という ペプチド設計用のプロトコルを指定。
-output→ 結果を保存する場所を指定するオプション。今回は、./output_run1を指定し、今いるフォルダの中にoutput_run1を作ってそこに保存させた。-num_designs→ 何個の設計を作るかを指定するオプション。今回は5個を指定。実際は、論文によると1~6万個ほどを指定し、そこから最適なものを探すのが良いそう。-budget→ 計算の強さ(探索の回数やコスト)を指定するオプション。今回は、テスト用に2を指定。大きくすると精度が上がるが、計算時間が増える。
最後に
皆様、いかがだったでしょうか。BolzgenはYAMLファイルの書き方に癖があるものの非常に多彩な設計やスコア評価を、一つのツールで一貫してできることが分かったかと思います。
RFPeptideと比較してどちらが高性能かは議論の余地があるものの、一つで構造生成、配列決定、評価までが出来るこのツールの便利さが実感できたかと思います。
これからの創薬でも利用されていきそうなこのツールを、ぜひ、皆様も実際に動かしてみてください!
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

