

デザインした環状ペプチドから候補を絞るには、docking評価の他にMDシミュレーションが欠かせません。本記事では、研究から実務レベルで広く利用されるMDシミュレーションツール「OpenMM」を用いた、タンパク質-ペプチド複合体のMDシミュレーションの実行方法とシミュレーション結果の定量評価の手法を具体的に解説していきます。
【この記事のまとめ】
環状ペプチドのデザイン後、静的なドッキング評価だけでは不十分な「結合の安定性」を、OpenMMを用いた分子動力学(MD)シミュレーションによって原子レベルで動的に検証する実践的な手法を解説します。
- 「動的な妥当性」の検証: Bolzgenなどで生成した静止構造が、溶液中での熱ゆらぎや溶媒環境下でも安定した相互作用を維持できるかを、物理法則に基づいてシミュレーションし、信頼性を判断できます。
- OpenMMによる高速・柔軟な解析: Pythonライブラリとして動作するOpenMMを活用し、GPUによる高速計算(CUDA)とスクリプト制御を組み合わせた、研究・実務レベルのMD実行フローを網羅。
- 5つの指標による定量評価: RMSD(構造のずれ)、RMSF(残基の揺れ)、水素結合数、最小距離、回転半径(Rg)を可視化し、ペプチドが解離せず結合様式が維持されているかを客観的に評価。
この記事を読み進めることで、専門的なMDエンジンの環境構築から、シミュレーション実行、そして解析結果のプロット作成までを一気通貫で習得でき、In silico創薬の精度を飛躍的に高めることが可能です。
Windows 11, WSL2(Ubuntu 20.04),bash
PCスペック
CPUメモリ: 16GB
GPU0: NVIDIA GeForce RTX 2070 SUPER(専用メモリ 8GB)
GPUドライバー: 581.29-desktop-win10-win11-64bit-international-dch-whql

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


MDシミュレーションとは?
In silicoペプチド創薬における分子動力学(MD)シミュレーションは、タンパク質–ペプチド複合体構造が「物理的に妥当で、本当に安定に存在しうるか」を検証するためのステップです。このステップでは、コンピューター上で分子を原子レベルで時間発展させることで、溶液中における分子の振る舞いを再現し、タンパク質とペプチドの結合が実際に安定して維持されるかを評価します。Bolzgenなどで生成した静的な構造モデルが単に見かけ上もっともらしい配置を示しているだけなのか、それとも熱ゆらぎや溶媒環境を考慮しても安定な相互作用を保つのかを、動的に観察することで解析できる点がMDの大きな特徴です。
本記事のワークフローでは、bolzgenによって直接生成された環状ペプチドと標的タンパク質の複合体構造のMDを評価するためにOpenMMを実施します。特にペプチドは柔軟性が高く、初期モデルから構造が緩和したり、相互作用が再編成されたりすることが多いため、MDなしでは結合の信頼性を十分に判断できません。複合体構造に対してMDシミュレーションを行うことで、その結合が維持されるのか、重要な相互作用が安定しているのか、あるいは結合様式が大きく変化するのかといった、静的モデルからは得られない重要な情報を把握することが可能になります。
OpenMMとは?
OpenMMは、「分子動力学(MD)シミュレーション」を簡単かつ高速に実行できるオープンソースソフトウェアです。GPUを利用した高速計算と、Pythonから直感的に操作できる点が特徴で、タンパク質やペプチド複合体のシミュレーションを比較的短いコードで実行できます。
MDシミュレーションでは、分子を構成する原子一つひとつに働く力(力場)を計算し、その力に基づいてニュートンの運動方程式を数値的に解くことで、時間とともに原子がどのように動くかを追跡します。OpenMMは、この「力の計算」と「時間発展の積分」を効率よく行う事が可能で、水分子やイオンを含めた現実に近い環境で分子の振る舞いを再現できます。
実際のシミュレーションは、①構造データの読み込み、②力場の設定、③水やイオンの追加、④エネルギー最小化、⑤温度・圧力を整える平衡化、⑥本番シミュレーションの実行、という流れで進みます。OpenMMではこれらをPythonスクリプトで順に設定できるため、プロトコルの理解と再現がしやすく、ドッキング後の複合体が溶液中で安定に存在できるかを評価するのに適しています。
本記事では、このOpenMMを用いてタンパク質・ペプチド複合体のMDシミュレーションをどのように準備し、実行するのか、さらにMD結果の定量的な確認方法を、初心者にも分かるように順を追って解説していきます。
GROMACSとの違い
| 項目 | GOMACS | OpenMM |
|---|---|---|
| 位置づけ | 伝統的かつ完成された定番MD実行エンジン | プログラマブルなMDライブラリ |
| 操作方法 | 設定ファイル(.mdp)+コマンドライン実行 | Python からスクリプト制御 |
| GPU設計 | CPUのみ または、GPU併用 | GPU前提 |
| カスタマイズ性 | やや限定的 | カスタム力場・独自ポテンシャル実装が容易 |
GROMACS と OpenMM は、いずれも高性能な分子動力学(MD)シミュレーションソフトウェアですが、設計思想と利用スタイルに明確な違いがあります。
GROMACSは、長年にわたり発展してきた成熟したMDエンジンであり、高速かつ安定した計算性能に定評があります。設定ファイル(.mdpなど)を用いてシミュレーション条件を定義し、コマンドラインから実行するスタイルが基本となります。そのため、大規模系のルーチン計算や再現性の高い標準的なMDワークフローにおいて非常に強力です。実験系研究者や計算化学分野での定番ツールとして広く使用されています。
一方、OpenMMは「完成された実行プログラム」というよりも、**「MD計算を構築するためのプログラマブルなライブラリ」**に近い位置づけです。Pythonから直接シミュレーションを組み立てる設計になっており、計算条件やアルゴリズムをコードレベルで柔軟に制御できます。このため、カスタム力場の導入、新規ポテンシャルの実装、機械学習モデルとの統合など、研究開発色の強い用途に適しています。
計算アーキテクチャの観点でも違いがあります。GROMACSはCPU並列化を基盤にしつつGPUアクセラレーションを統合する設計であるのに対し、OpenMMは当初からGPU活用を前提に設計されており、CUDAやOpenCLを利用した効率的なGPU計算に強みを持ちます。
まとめると、GROMACSは「高性能で完成度の高いMD実行エンジン」、OpenMMは「柔軟性と拡張性を重視したMD開発フレームワーク」と捉えると理解しやすいでしょう。長年GROMACSを使用してきた研究者にとって、OpenMMは操作感そのものが異なり、よりプログラミング主導でMDを設計することを前提にしたツールであると言えます。
環境構築
まずはこちらを参考に環境構築から行っていきます。今回もWindowsのWSLを利用していきます。WSLの利用方法はこちらの記事に記載してあるので、そちらを参考にしてください。
今回のツールはGPUを利用するツールのため、まずはこちらのサイトでお使いのPCに搭載されているGPUに 対応したドライバ(Windows11用。Linux用ではありません)をDLして、Windows 上でインストーラを実行し、再起動して有効化してください。また、どのドライバーが良いのか分からない場合は、こちらのNDIVIA appをインストールすると最適なドライバーをインストールしてくれるようです(既にインストール済みの場合は不要です。)
次に、WSLのターミナルで以下のコードを実行して、環境構築をしていきます。
ターミナルを開き、以下を実行してください。
# ① 環境をconda-forgeのみで作成
conda create -n peptide-md -c conda-forge python=3.11 -y
conda activate peptide-md
# ② 依存解決を高速化
conda install -n peptide-md -c conda-forge mamba -y
# ③ CUDA対応OpenMM + PDBFixer をインストール
mamba install -c conda-forge \ openmm \ cuda-version=12 \ pdbfixer \ mdtraj \ matplotlib \ numpy \ -yこれでWSL上にOpenMM環境が構築されました!
複合体構造の修正とOpenMMでのMDの実行
今回は以下の記事で生成した環状ペプチドのMDを行ってみます。
【環状ペプチド】Bolzgenを使ったDe novo環状ペプチドの生成【In silico創薬】 – LabCode
以下のfinal_2_designの中にあるCIFファイルの中からMDにかけたいファイルを決め、その名前をProtein_Peptide.cifに変更してください(PDBファイルの場合でも大丈夫です。)。
WSLターミナルを開き、
mkdir -p ~/md_run
cd ~/md_runと入力し、MD作業用のディレクトリを作成・移動します。WSL側に用意することでシミュレーションを高速に実行できます。そして、以下のコマンドでこのディレクトリ内に、MDシミュレーションにかけたい複合体のPDBファイルを移動させておきます。
cp /mnt/c/Users/<Windowsのユーザー名>/Documents/bolzgen/output_run1/final_ranked_designs/final_2_designs/Protein_Peptide.cif .こちらのコマンドは、CIFファイル(またはPDBファイル)の保存場所に合わせて適宜変更して下さい。
ここまで終わったら以下を実行して下さい。
# 複合体構造の修正(立体構造ファイル名や拡張子などは適宜変更して下さい)
pdbfixer Protein_Peptide.cif \ --add-atoms=all \ --ph=7.0 \ --output=fixed.pdb
# nanoというファイル編集ツールを開く
nano run_openmm.py実行するとファイル編集画面が開くと思います。
そこに以下をコピペして下さい。
from openmm.app import *
from openmm import *
from openmm.unit import *
from sys import stdout
print("Loading structure...")
pdb = PDBFile("fixed.pdb")
print("Loading force field...")
forcefield = ForceField("amber14-all.xml", "amber14/tip3p.xml")
modeller = Modeller(pdb.topology, pdb.positions)
print("Adding solvent...")
modeller.addSolvent( forcefield, padding=1.0*nanometers, ionicStrength=0.15*molar
)
print("Saving solvated system (system.pdb)...")
PDBFile.writeFile( modeller.topology, modeller.positions, open("system.pdb", "w")
)
print("Creating system...")
system = forcefield.createSystem( modeller.topology, nonbondedMethod=PME, nonbondedCutoff=1*nanometer, constraints=HBonds
)
print("Integrator...")
integrator = LangevinIntegrator( 300*kelvin, 1/picosecond, 0.002*picoseconds
)
integrator.setRandomNumberSeed(42)
print("Using GPU...")
platform = Platform.getPlatformByName("CUDA")
properties = {"Precision": "mixed"}
simulation = Simulation( modeller.topology, system, integrator, platform, properties
)
simulation.context.setPositions(modeller.positions)
print("Minimizing energy...")
simulation.minimizeEnergy()
print("Equilibrating (100 ps)...")
simulation.context.setVelocitiesToTemperature(300*kelvin)
simulation.step(50000)
print("Setting reporters...")
simulation.reporters.append( DCDReporter("traj.dcd", 1000)
)
simulation.reporters.append( StateDataReporter( "log.txt", 1000, step=True, temperature=True, potentialEnergy=True, density=True, speed=True )
)
print("Production run (5 ns)...")
simulation.step(2500000)
print("Done.")コピペ後には、キーボードでCtrl+O → Enter → Ctrl+Xを入力して、ファイルの保存を行って下さい。このファイルがOpenMMを実行する為のpythonファイルになっています。
最後にこのpythonファイルをWSL上で実行します。これでopenMMによるMDが実行されます。
(数十分から数時間かかります。)
python run_openmm.pyMD結果の確認
今回はWSL上で実行したので、実行結果のファイルはWSL上に保存されています。
まずは以下のコマンドで、実行結果ファイルがあるかを確認しましょう。
ls -lhこのコマンドで以下のようになっていればMDは成功です!
(peptide-md) xx@XX:~/md_run$ ls -lh total 1.4G -rwxr-xr-x 1 xx xx 97K Feb 27 15:17 Protein_Peptide.cif -rw-r--r-- 1 xx xx 170K Feb 27 15:18 fixed.pdb -rw-r--r-- 1 xx xx 335K Feb 27 16:19 log.txt -rw-r--r-- 1 xx xx 1.4K Feb 27 15:19 run_openmm.py
-rw-r--r-- 1 xx xx 1.8M Feb 27 16:19 system.pdb
-rw-r--r-- 1 xx xx 1.3G Feb 27 16:19 traj.dcdlog.txtには実行ログが、traj.dcdとsystem.pdbにはMD計算の結果が入っています。
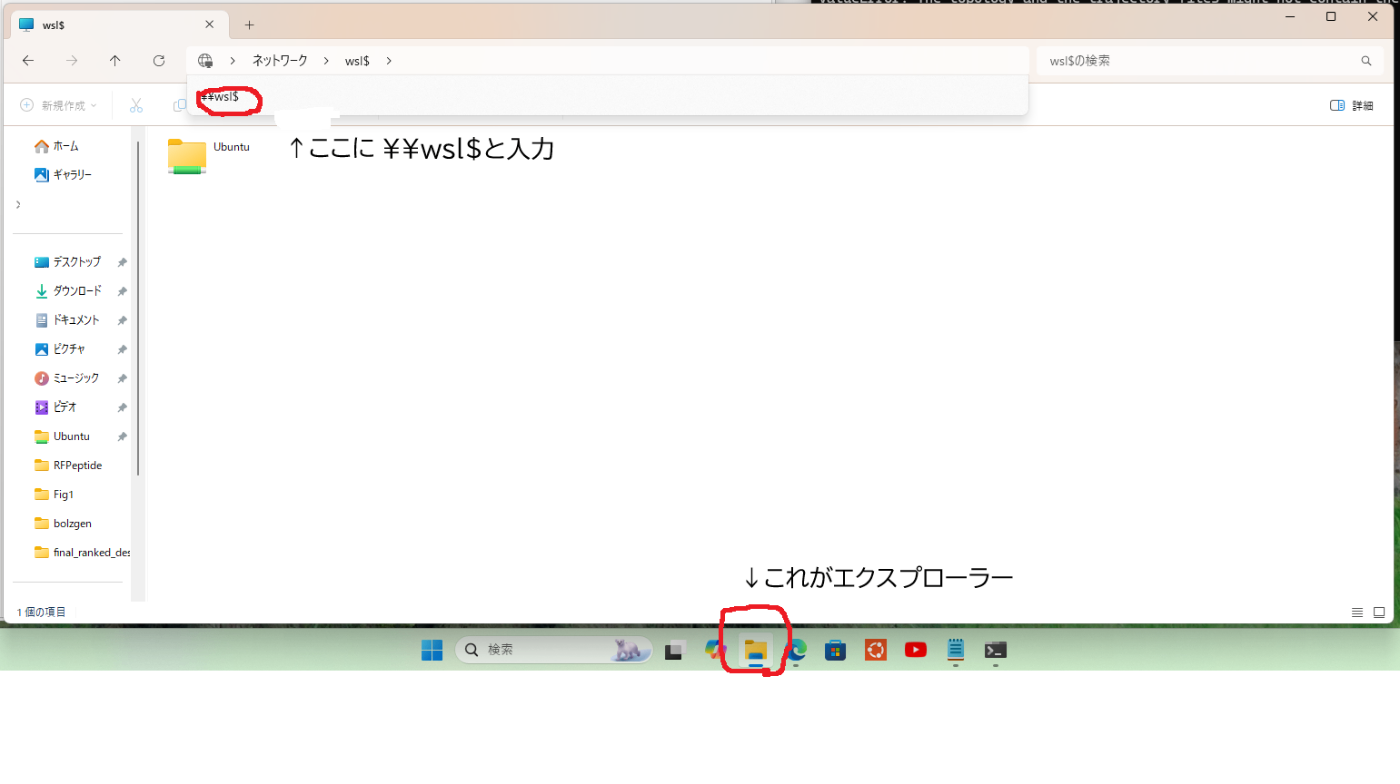
実はWSL上のファイルは、Windows端末から直接確認することができます。
エクスプローラーのアドレスバー(下の画像を参照)に\\wsl$ と入力すると、これらのWSL上のファイルにWindows上のファイルかのようにアクセスすることが可能です。
Ubuntu→home→ユーザー名 →md_run(今回作成した環境名)と進んでいくと、これらの結果ファイルにアクセスできますので、この方法で確認するのもいいでしょう。
次に以下のコマンドで、pythonで動くmdtrajというパッケージを使って、このtraj.dcdとsystem.pdbの中身を分析していきます。これらのファイルには、時間変化ごとの立体構造の軌跡が入っているのですがそのままでは結果を把握できないので、この軌跡の情報を分析して定量的に評価するために、以下の5つの要素をプロットで可視化していきます。
- RMSD
- 全体構造のずれ
- → システムが安定しているか確認
- RMSF
- 各残基の揺れ
- → 柔らかい部分・不安定部位の特定
- 水素結合(H-bond)
- タンパク–ペプチド間の水素結合数
- → 相互作用の安定性評価
- 最小距離(Minimum Distance)とコンタクト数
- 両者の最短原子間距離や接触の数
- → 解離していないか確認
- 回転半径(Radius of Gyration, Rg)
- 構造の広がり具合
- → 折りたたみ状態・崩壊の有無を確認
これらをプロットすることで、
- 構造が安定しているか
- ペプチドが解離していないか
- 結合様式が維持されているか
- 特定領域が過度に揺らいでいないか
といった点を総合的に判断できるようになります。
MDの結果ファイルからこれらの情報を抽出するためのpythonコードを、nanoを用いて作成します。まずは、以下のコマンドを実行してください。
# nanoというファイル編集ツールを開く
nano analyze_md.py実行するとファイル編集画面が開くと思います。
そこに以下をコピペして下さい。
#!/usr/bin/env python3
import mdtraj as md
import numpy as np
import matplotlib.pyplot as plt
# ===============================
# 0. 設定
# ===============================
EQUIL_TIME_NS = 0.1 # 平衡化の時間(単位ns)
CONTACT_CUTOFF = 0.4 # nm (4.0 Å)
print("Loading trajectory...")
traj = md.load("traj.dcd", top="system.pdb")
# ===============================
# 1. PBC補正 & センタリング
# ===============================
print("Applying PBC correction and centering...")
traj.center_coordinates()
traj = traj.image_molecules()
traj = traj[traj.time / 1000 >= EQUIL_TIME_NS] # 平衡化の時間分差し引いてプロットをスタート
start_time = EQUIL_TIME_NS
time_ns = start_time + np.arange(traj.n_frames) * 0.002
# ===============================
# 2. Atom selection
# ===============================
# Cα (RMSD/RMSF用)
protein_ca = traj.topology.select("chainid 0 and name CA")
peptide_ca = traj.topology.select("chainid 1 and name CA")
complex_ca = traj.topology.select("(chainid 0 or chainid 1) and name CA")
# 重原子 (Contacts/MinDist/Rg用)
protein_heavy = traj.topology.select("chainid 0 and not element H")
peptide_heavy = traj.topology.select("chainid 1 and not element H")
complex_heavy = traj.topology.select("(chainid 0 or chainid 1) and not element H")
# 全原子セット (Hbond用判定用)
protein_atoms = set(traj.topology.select("chainid 0"))
peptide_atoms = set(traj.topology.select("chainid 1"))
# ===============================
# 3. アラインメント
# ===============================
print("Aligning trajectory...")
traj.superpose(traj, 0, atom_indices=protein_ca)
# ===============================
# 4. RMSD (Complex追加)
# ===============================
print("Calculating RMSD...")
rmsd_protein = md.rmsd(traj, traj, 0, atom_indices=protein_ca) * 10
rmsd_peptide = md.rmsd(traj, traj, 0, atom_indices=peptide_ca) * 10
rmsd_complex = md.rmsd(traj, traj, 0, atom_indices=complex_ca) * 10
plt.figure(figsize=(8, 5))
plt.plot(time_ns, rmsd_protein, label="Protein (GABARAP)")
plt.plot(time_ns, rmsd_peptide, label="Peptide (Cyclic)")
plt.plot(time_ns, rmsd_complex, label="Complex", color="black", linewidth=1.5)
plt.xlabel("Time (ns)")
plt.ylabel("RMSD (Å)")
plt.legend()
plt.title("Cα RMSD (10ns Full Run)")
plt.grid(True, linestyle='--', alpha=0.6)
plt.savefig("rmsd_ca_10ns.png")
plt.close()
# ===============================
# 5. RMSF (連続プロット)
# ===============================
print("Calculating RMSF...")
rmsf_protein = md.rmsf(traj, traj[0], atom_indices=protein_ca) * 10
rmsf_peptide = md.rmsf(traj, traj[0], atom_indices=peptide_ca) * 10
prot_res_count = len(protein_ca)
res_idx_protein = np.arange(prot_res_count)
res_idx_peptide = np.arange(len(peptide_ca)) + prot_res_count
plt.figure(figsize=(10, 5))
plt.plot(res_idx_protein, rmsf_protein, label="Protein", color="blue")
plt.plot(res_idx_peptide, rmsf_peptide, label="Peptide", color="orange")
plt.axvline(x=prot_res_count - 0.5, linestyle="--", color="red", label="Boundary")
plt.xlabel("Residue Index")
plt.ylabel("RMSF (Å)")
plt.legend()
plt.title("Cα RMSF (10ns)")
plt.savefig("rmsf_ca_10ns.png")
plt.close()
# ===============================
# 6. H-bonds (Inter-molecular only)
# ===============================
print("Calculating Inter-molecular H-bonds...")
hbonds_raw = md.wernet_nilsson(traj, periodic=True)
hbond_counts = []
for frame_bonds in hbonds_raw: count = 0 for a, h, d in frame_bonds: if (a in protein_atoms and d in peptide_atoms) or \ (a in peptide_atoms and d in protein_atoms): count += 1 hbond_counts.append(count)
plt.figure(figsize=(8, 5))
plt.plot(time_ns, hbond_counts, color="green")
plt.xlabel("Time (ns)")
plt.ylabel("Number of H-bonds")
plt.title("Protein–Peptide Inter-molecular H-bonds")
plt.savefig("hbonds_10ns.png")
plt.close()
# ===============================
# 7. Heavy Atom Min Distance & Contacts (Inter-molecular)
# ===============================
print("Calculating Heavy Atom distances and contacts (Inter-molecular)...")
# ペアをタンパク質重原子 vs ペプチド重原子に限定することでメモリを節約
heavy_pairs = []
for i in protein_heavy: for j in peptide_heavy: heavy_pairs.append((i, j))
# 分子間重原子ペアのみの距離計算
distances = md.compute_distances(traj, heavy_pairs, periodic=True)
# 最小距離 (Å)
min_dist = distances.min(axis=1) * 10
# コンタクト数 (0.4nm以内の重原子ペア数)
contact_count = (distances < CONTACT_CUTOFF).sum(axis=1)
plt.figure(figsize=(8, 5))
plt.plot(time_ns, contact_count, color="purple")
plt.xlabel("Time (ns)")
plt.ylabel("Number of Heavy Atom Contacts")
plt.title("Protein–Peptide Heavy Atom Contacts")
plt.savefig("contacts_heavy_10ns.png")
plt.close()
plt.figure(figsize=(8, 5))
plt.plot(time_ns, min_dist, color="red")
plt.xlabel("Time (ns)")
plt.ylabel("Min Heavy Atom Distance (Å)")
plt.title("Protein–Peptide Minimum Heavy Atom Distance")
plt.savefig("mindist_heavy_10ns.png")
plt.close()
# ===============================
# 8. Radius of Gyration (Complex)
# ===============================
print("Calculating Rg for Complex...")
# protein_heavy ではなく complex_heavy を使用してスライス
complex_traj = traj.atom_slice(complex_heavy)
rg_complex = md.compute_rg(complex_traj) * 10
plt.figure(figsize=(8, 5))
plt.plot(time_ns, rg_complex, color="brown")
plt.xlabel("Time (ns)")
plt.ylabel("Rg (Å)")
# タイトルも Complex に変更
plt.title("Complex Radius of Gyration (Heavy Atoms)")
plt.savefig("rg_complex_10ns.png")
plt.close()
print("\nDone! All analysis files generated.")コピペ後には、先ほどと同様にキーボードでCtrl+O → Enter → Ctrl+Xを入力して、ファイルの保存を行って下さい。このファイルがtraj.dcdファイル等を分析するためのpythonファイルになます。
最後にこのpythonファイルをWSL上で実行します。これでopenMMによるtraj.dcdファイル等の分析が実行されます。
python analyze_md.py実行するとつの画像ファイルができているはずです。先ほどのwindowsのエクスプローラーから確認してみてください。
以下の様になっていれば成功です(各画像の解釈は後程、解説します)。
コードの解説
上に書いたソースコードの解説をしていきます。
先ずは、①環境構築のコマンドから解説をします。
# ① 環境をconda-forgeのみで作成
conda create -n peptide-md -c conda-forge python=3.11 -y
conda activate peptide-md
# ② 依存解決を高速化
conda install -n peptide-md -c conda-forge mamba -y
# ③ CUDA対応OpenMM + PDBFixer をインストール
mamba install -c conda-forge \ openmm \ cuda-version=12 \ pdbfixer \ mdtraj \ matplotlib \ numpy \ -y先ず以下で、condaでpeptide-md という環境を用意します。
conda create -n peptide-md -c conda-forge python=3.11 -y conda activate peptide-md
さらに、以下のコマンドでconda-forgeを使って、mambaをインストールします。mamba はcondaの高速版で、openMMはインストールに時間がかかるので、今回はこちらを利用します
conda install -n peptide-md -c conda-forge mamba -y
さらに、以下のコマンドで必要なパッケージをインストールします。
mamba install -c conda-forge \ openmm \ cuda-version=12 \ pdbfixer \ mdtraj \ matplotlib \ numpy \ -y
それぞれのパッケージには以下のような役割があり、-y でユーザーが承認しなくとも自動でインストールされるように設定してあります。
👉 openmm → MDエンジン
👉 cuda-version=12 → GPUを使えるようにする
👉 pdbfixer → 構造修正
👉 mdtraj →MD結果の分析
👉 matplotlib・numpy → 可視化、数値計算
次に、②複合体構造の修正とOpenMMでのMDの実行を解説します。
先ずは、pdbfixerの実行部分について解説します。
# 複合体構造の修正
pdbfixer Protein_Peptide.cif \ --add-atoms=all \ --ph=7.0 \ --output=fixed.pdb
# nanoというファイル編集ツールを開く
nano run_openmm.pyここでは、pdbfixer を使って、Bolzgenで生成した Protein_Peptide.cif(元の構造ファイル)を修理しています。
オプションの意味は以下。
-add-atoms=all→ 足りない原子(特に水素)を全部追加-ph=7.0→ pH7(生理条件)で水素の付き方を決める-output=fixed.pdb→ 修理後の構造を fixed.pdb という名前で保存
なぜこの処理が必要かというと、Bolzgenの生成した構造ファイルは、原子モデルとしては不完全な状態 (水素原子が入っていない場合や一部の原子が欠けている、電荷状態が決まっていないといった状態)であることが多く、そのままMDを行うと物理計算が出来ないことがあるためです。今回は、水素原子とpHの設定を行い、より“完全な原子モデル”に整えたのちに、MD計算を行っていきます。
また。それと同時にnano というテキストエディタでrun_openmm.py というpythonファイルを作成しています。openMMはオプションコマンドではなく、pythonファイルでMDの詳細設定を指示する必要があるためです。
では、nano で作成したrun_openmm.py の中身を解説していきます。これはopenMMの実行ファイルとなっており、「タンパク質+ペプチドを水の中に入れて、300Kで10ナノ秒動かす」という分子シミュレーションを行うように指定してあります。
以下で詳しく解説します。
🔹 ① ライブラリの読み込み
from openmm.app import * from openmm import * from openmm.unit import * from sys import stdout
👉 OpenMMを使うためのパッケージを読み込んでいます。 unit は nm や kelvin などの単位を扱うためのもの。
🔹 ② 構造ファイルの読み込みpdb = PDBFile('fixed.pdb')
👉 fixed.pdb(PDBFixerで整えた構造)を読み込んでいます。 これが計算の出発点。
🔹 ③ 力場(force field)の読み込みforcefield = ForceField('amber14-all.xml', 'amber14/tip3p.xml')
👉 分子にかかる力のルールを指定しています。
amber14-all.xml → タンパク質用
tip3p.xml → 水分子用
つまり 「タンパク質と水をどう計算するか」を決めています。
🔹 ④ 溶媒を追加modeller.addSolvent(...)
👉 タンパク質の周囲に溶媒(今回は水)を入れています。
padding=1.0 nm → 周囲1nmの水の層
ionicStrength=0.15 M → 生理食塩水くらいの塩濃度
現実の体内環境に近づけています。
🔹 ⑤ 解析用トポロジー保存PDBFile.writeFile(...)
結果の保存方法を指定。
traj.dcd は座標だけ
system.pdb が原子定義を持つ
🔹 ⑥ システム作成system = forcefield.createSystem(...)
👉 ここで「物理計算モデル」を作っています。
PME → 長距離の静電相互作用を正確に計算
constraints=HBonds → 水素結合の長さを固定(高速化)
ここが“物理エンジン”部分。
🔹 ⑦ 温度制御設定integrator = LangevinIntegrator(...)
👉 300K(約27℃)で計算する設定。
300 K → 温度
1/picosecond → 摩擦係数
0.002 ps → 2 fsステップ
2フェムト秒刻みで時間を進めます。
🔹 ⑧ GPUを使う設定platform = Platform.getPlatformByName('CUDA')
👉 NVIDIA GPUを使う指定。 GPUで高速計算します。
🔹 ⑨ エネルギー最小化simulation.minimizeEnergy()
👉 ぶつかっている原子を少し整えます。 「初期構造のほぐして、自然な状態にする」。
🔹 ⑩ 平衡化(100 ps)simulation.context.setVelocitiesToTemperature(300*kelvin) simulation.step(50000)
2 fs × 50,000 = 100 ps
👉 シミュレーション計算で少しだけ時間を経過させ、分子構造を平衡化(安定)させる。
🔹 ⑪ 出力設定DCDReporter('traj.dcd', 1000)
👉 1000ステップごとに座標保存 → traj.dcd(軌道ファイル)
StateDataReporter('log.txt', ...)
👉 温度・エネルギーなどを保存 → log.tx
🔹 ⑫ 本番計算(10 ns)simulation.step(5000000)
2 fs × 5,000,000 = 10 ns
👉 実際の分子運動シミュレーション。
最後に③MD結果の確認について解説します。
ここでも同様にnano analyze_md.py というコマンドで、analyze_md.py というpythonファイルを作成しています。analyze_md.py の中身は以下で詳しく解説します。
🔹 ① ライブラリの読み込み
import mdtraj as md import numpy as np import matplotlib.pyplot as plt
mdtraj: MD解析のデファクトスタンダード。軌跡の読み込み、距離計算、水素結合判定などを高速に行います。
numpy: 配列計算の基礎。計算された距離や時間の単位変換、統計処理に使用します。
matplotlib: データの可視化。
🔹 ② 軌跡ファイルの読み込み
traj = md.load("traj.dcd", top="system.pdb")traj.dcd: 座標のタイムコースデータ。原子が「いつ、どこにいたか」の記録です。system.pdb: 座標に意味を与える構造情報。「どの原子が、どの残基に属し、どのチェーンか」を紐付けます。
🔹 ③ PBC補正と平衡化部分の除外
traj.center_coordinates() traj = traj.image_molecules()PBC補正 (center / image): 周期境界条件により箱の外へ飛び出した分子を箱の中に引き戻し、タンパク質を中心に据えます。これをしないと、分子が瞬間移動したような異常なグラフになります。
traj = traj[traj.time / 1000 >= EQUIL_TIME_NS] start_time = EQUIL_TIME_NS
平衡化の除外: シミュレーション開始直後の不安定な初期状態を解析から捨てることで、平衡化後の安定状態の値を解析に利用します。
time_ns = start_time + np.arange(traj.n_frames) * 0.002
時間軸の再構成: 0.002ns (2ps) ごとに書き込まれたフレームとプロットの時間軸(横軸)を合わせます。
🔹 ④ 解析対象の選択 (Selection)
`# 重原子 (Contacts/MinDist/Rg用) protein_ca = traj.topology.select(“chainid 0 and name CA”) peptide_ca = traj.topology.select(“chainid 1 and name CA”) complex_ca = traj.topology.select(“(chainid 0 or chainid 1) and name CA”)
全原子セット (Hbond用判定用)
protein_heavy = traj.topology.select(“chainid 0 and not element H”) peptide_heavy = traj.topology.select(“chainid 1 and not element H”) complex_heavy = traj.topology.select(“(chainid 0 or chainid 1) and not element H”)`
役割: 「システム全体(溶媒含む)」から「解析したい部分」だけを抽出します。
CA原子のみ : アミノ酸の中心のCのみを抜き出す。骨格の動きを見るため、RMSD/RMSFに使用。
Heavy Atoms (重原子)のみ: 水素以外の全原子を選択。密着度や接触を詳しく見るためContacts / MinDistで利用。
これにより、不要な原子(水やイオン)を計算から外すことで、メモリ消費を抑え、計算を高速化します。
🔹 ⑤ 重ね合わせ (Alignment)
traj.superpose(traj, 0, atom_indices=protein_ca)役割: 全フレームのタンパク質を第1フレームにピッタリ重ね合わせます。MD中に起きた分子自体の「回転」や「拡散移動」によるノイズを取り除きます。これにより、グラフに現れる変化が「純粋な構造変化」であることを保証します。
🔹 ⑥ RMSD(安定性)の解析
rmsd_complex = md.rmsd(traj, traj, 0, atom_indices=complex_ca) * 10RMSD: 「初期構造からのズレ」を定量化します。これが一定値で横ばいになれば、構造が安定した(平衡化した)と判断できます。複合体全体、タンパク質単体、ペプチド単体の3つを算出します。Ca原子のみで計算します。
単位変換: * 10 をすることで、ナノメートルから一般的な単位であるÅ(オングストローム )に変換しています。
🔹 ⑦ RMSF(柔軟性)の解析
rmsf_protein = md.rmsf(traj, traj[0], atom_indices=protein_ca) * 10役割: 残基ごとの「揺れ」の平均を算出します。ペプチドとの結合部位が固定されているかを確認するのに有効です。Ca原子のみで計算します。
🔹 ⑧ H-bonds(相互作用)の解析
hbonds_raw = md.wernet_nilsson(traj, periodic=True)
H-bonds: 経過時間ごとのペプチドとタンパク間の水素結合数をカウントします。ペプチドとタンパクの全原子を対象に計算します。
🔹 ⑨ 接触と距離の解析 (Heavy Atom Min Distance & Contacts)distances = md.compute_distances(traj, heavy_pairs, periodic=True) min_dist = distances.min(axis=1) * 10 contact_count = (distances < CONTACT_CUTOFF).sum(axis=1)
MinDist: 分子間の最短距離。ペプチドがポケットから離れていないか、あるいは衝突(貫通)していないかを確認します。
Contacts: 分子間で4.0 Å 以内に接近した原子ペアの数を経過時間ごとにカウントします。ペプチドがどれだけ「面」でタンパク質を捉えているかを示します。ペプチドとタンパクの重原子間のペアのみで計算を行います。
🔹 ⑩ 構造のコンパクトさの解析 (Radius of Gyration)
rg = md.compute_rg(protein_traj) * 10
Rg (回転半径): 「分子の広がり具合」。タンパク質がまとまっているか、あるいは広がっているかを数値化します。構造の崩壊などをチェックできます。複合体の重原子を対象に計算。
以上を踏まえて、今回作成されたプロットを確認していきます。
- RMSD(構造のずれ)
rmsd_ca_10ns.pngを開くと以下の様になっていました。
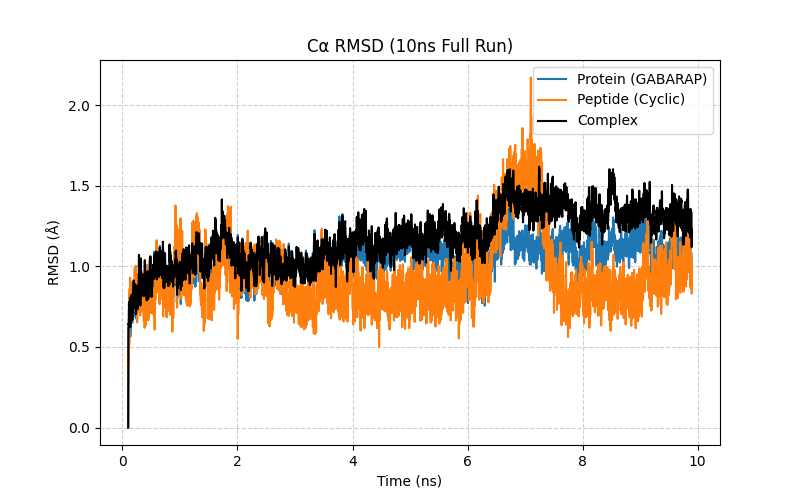
見方:初期構造からどれだけ構造が変化したかを時間で追跡しています。値が小さいほど初期構造に近く、安定しています。一般には振幅が 0.5Å 以内に収まっていれば「安定(Stable)」と記述できます。
今回の解釈:
-Protein (青): 1.2Å 程度で完全に一定です。これは GABARAP タンパク質自体が計算中に壊れず、安定していたことを意味します。
-Peptide (オレンジ): 7ns 付近で「跳ね」がありますが、その後すぐに 1.0Å 前後に戻っています。これは**「ポケットの中で少し動いたが、外れずに安定な位置に収まり直した」**ことを示しており、結合が強固であることを示唆します。
-Complex (黒): 全体として 1.5Å 以下に収まっており、複合体としての一体感が非常に高いです。
- RMSF(残基ごとの揺れ)
rmsd_ca_10ns.pngを開くと以下の様になっていました。
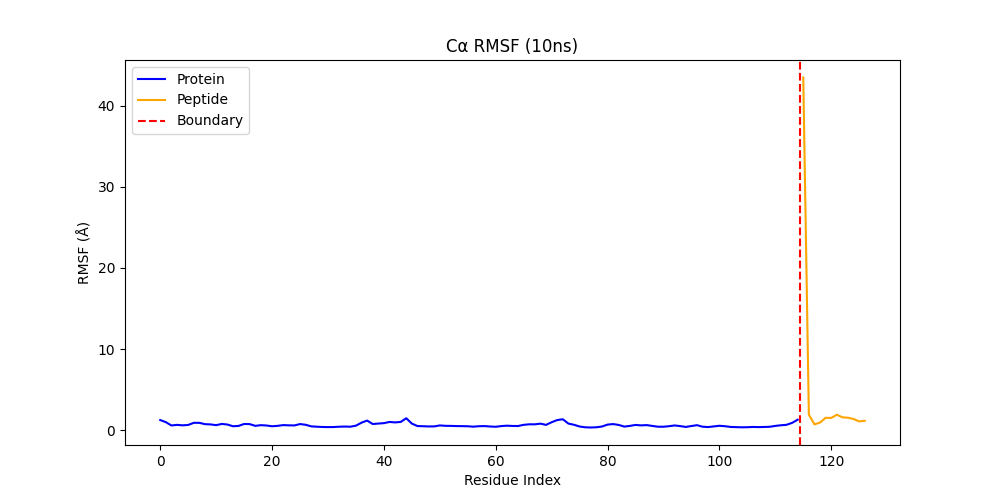
見方: 各残基が平均構造からどれだけ揺れたかを棒グラフで示しています。値が高いほどその残基は柔軟に動いています。赤い点線がタンパク質とペプチドの境界です。
今回の解釈:
-Protein (青): 低い値で静止しています。
-Peptide (オレンジ): 境界線のすぐ右側(ペプチドの端)にある 40Å を超える巨大なスパイクがある。
このスパイクは「ペプチドの特定の場所(おそらく端の数残基)」が激しく動いていることを示しています。しかし、その後の残基は 2Å 以下で安定しています。ペプチドの『核』となる部分はガッチリ固定されているが、端っこの部分だけが自由奔放に動いているという、結合モデルに見えます。しかし、40Å 越えはかなり高い印象です。
- 水素結合解析
hbonds_10nsを開くと以下の様になっていました。
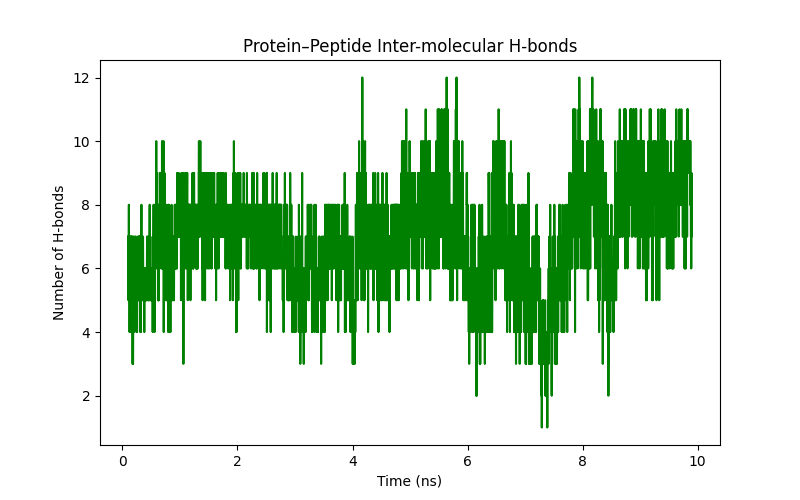
見方: タンパク質とペプチドを繋ぐ架け橋となる水素結合の数を経過時間ごとにプロットしています。水素結合が多いと両者の結合は強固であり、数やその時間当たりの変化は結合強度の指標になります。今回の解釈: 常時 6〜10本 という、非常に多い数の水素結合が維持されています。特に 8ns 以降で数が安定して増えているように見えます。
- 最小距離&コンタクト解析
mindist_heavey_10ns.pngを開くと以下の様になっていました。
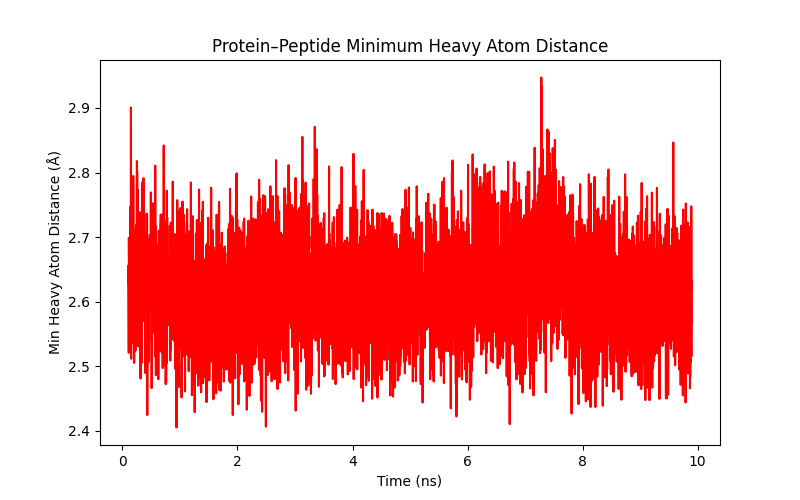
見方: 両分子の原子が最も近づいている距離(最小距離)です。2.5~3.2Å 程度:だと安定した水素結合や強固な塩橋が形成されている可能性が高い。
今回の解釈: 常に 2.5Å 〜 2.9Å 付近を推移しています。これは水素結合の理想的な距離であり、10ns の間、ペプチドがポケットから離れていない(浮いていない)ことを示しています。
cantacts_heavey_10ns.pngを開くと以下の様になっていました。
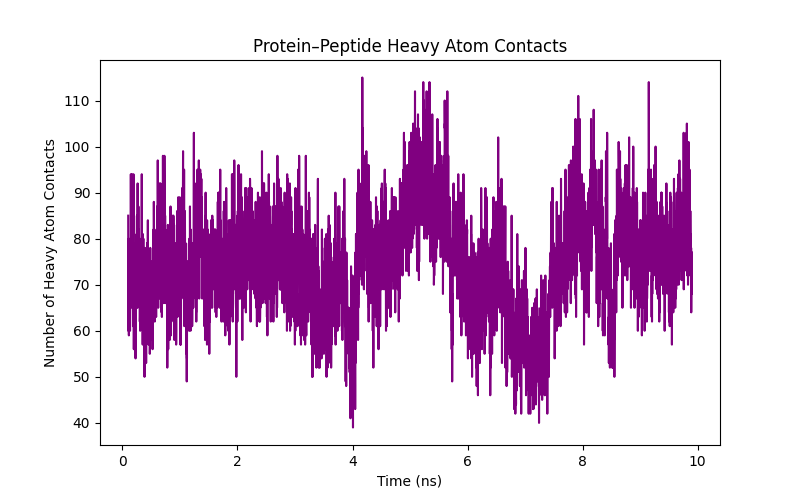
見方: 0.4nm 以内に接近している原子ペア(コンタクト)の数です。密着度を表します。一定であると結合が継続しているとみなせます。
今回の解釈: 平均して 70〜90 個程度の接触を維持しているように見えます。7.5ns 付近で一時的に下がっていますが(RMSD の跳ねと連動)、その後すぐに回復しています。
- 回転半径(Radius of Gyration)
rg_complex_10ns.pngを開くと以下の様になっていました。
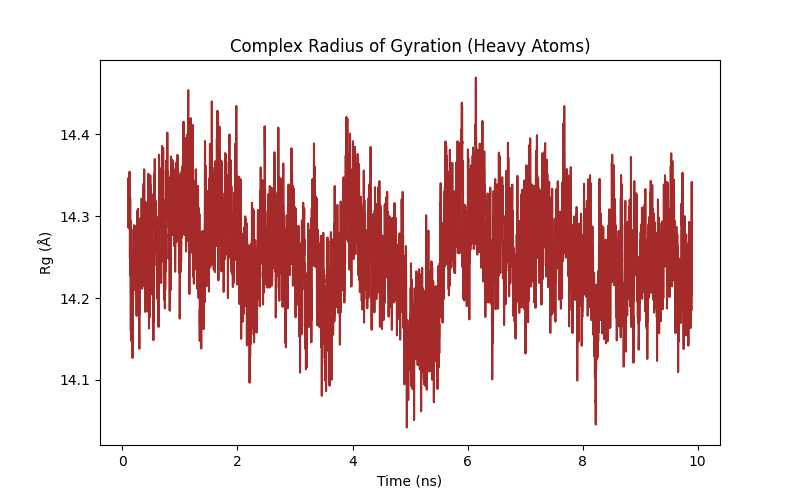
見方: 複合体の「コンパクトさ(凝縮度)」です。シミュレーション期間を通じて一定、もしくは微減する場合、「コンパクトな複合体を維持している」と解釈します。
今回の解釈: 14.0Å 〜 14.4Å の非常に狭い範囲で上下しています。これは、タンパク質がペプチドを抱え込んだまま、ふらつくことなくガッチリした形を保っている証拠です。
最後に
以上がタンパク質と環状ペプチドとのMDシミュレーションのやり方になります。計算時間の長さや行程の複雑さはかなりヘビーでしたが、いかがでしたでしょうか。基本的にはタンパク質のみのMDと大差ないので、慣れている方は問題なくできたかと思います。
結果としては5つの解析すべてが一貫して、10 ns のシミュレーションを通じてタンパク質-ペプチド複合体は比較的安定に結合を維持していることを示していました。ペプチドはタンパク質より柔軟に動いていますが、水素結合・コンタクト数・最小距離のいずれも解離を示唆する変化はなく、安定した複合体として振る舞っています。
今回はMDの長さは10nsでしたが、論文などでは100ns以上で行う場合も多く、シミュレーションの長さや溶媒の種類、温度の条件などは目的に合わせて調節してみてください。また平衡化にももう少し時間をかけてもいいかもしれません。
ぜひマスターしてより高度なin silco創薬をマスターしてみてください!
参考文献
Eastman, P., et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Computational Biology (2017). https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005659
OpenMM公式サイト: https://openmm.org/
GitHub repository: https://github.com/openmm/openmm
License: MIT License https://github.com/openmm/openmm/blob/master/docs-source/licenses/Licenses.txt
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!
