

本記事では、mobiusというペプチド最適化ツールとベイズ最適化(Bayesian Optimization, BO)を組み合わせ、すでにアミノ酸変異によって結合力を予測したPSSM(Position-Specific Scoring Matrix)を用いてMHCクラスI(HLA-A*02:01)に対する結合親和性を向上させるペプチド配列を効率よく探索する方法を紹介します。
PSSMマトリックスを使用した高速な評価により、各ペプチドの結合スコア(pIC50)を取得し、その結果を基にベイズ最適化を実行することで、最も結合親和性の高いペプチド配列を見つけ出します。
【この記事のまとめ】
バイオインフォマティクスや創薬研究に携わるエンジニア・研究者に向けて、Pythonパッケージ「mobius」とベイズ最適化を活用し、標的タンパク質に対して高い結合親和性を持つペプチド配列を効率的に探索・設計する手法を解説します。
- 膨大な探索空間の克服:5000億通り以上(9残基の場合)存在するペプチド配列の組み合わせから、ベイズ最適化(BO)を用いて「賢く」次候補を選別し、実験・計算コストを劇的に削減する方法を習得できます。
- 実践的なモデリングフロー:アミノ酸変異による結合力予測(PSSM)とmobiusを組み合わせ、初期ライブラリの生成(アラニンスキャニング等)からDMTサイクル(設計・作成・試験)の自動化までのプロセスを詳述。
- 高度なカスタマイズ性:HELM記法によるエンコード、分子フィンガープリント(MAP4)を用いたガウス過程モデルの構築など、非標準アミノ酸や複雑な構造制約にも対応可能な最新の設計プロトコルを紹介。
この記事を読むことで、限られたリソースで最適解にたどり着く「データ駆動型ペプチド創薬」の具体的な実装スキルを身につけることができます。
動作検証済み環境
Mac M1, Sequoia 15.6

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


mobiusとは?
mobiusは、ベイズ最適化(Bayesian Optimization, BO)を用いてペプチド配列を最適化するためのPythonパッケージです。完全自動化されたDesign-Make-Test(DMT)サイクルの中で、ペプチドの設計と最適化を支援します。
mobiusの主な特徴は以下の通りです。
- 単一または複数の目的関数の最適化(制約条件付き)
- 線形および非線形ペプチド配列のサポート(マクロ環状、ラッソ、分岐など)
- 非標準アミノ酸残基と修飾のサポート
- デザインプロトコルによる配列最適化のカスタマイズ
- 独自の分子表現(フィンガープリント、グラフカーネル、GNNなど)の拡張が容易
- 他のツールとの統合(ドッキング、pyRosetta、Damietta、AlphaFoldなど)
今回は、mobiusのチュートリアルを参考にしつつ、PSSMマトリックスを使用した高速な評価を行う実装を行います。
MHCクラスI(HLA-A*02:01)に対する結合親和性を最適化するペプチド配列を、初期のリードペプチドからアラニンスキャニングとホモログスキャニングで96個のペプチドを生成し、それぞれをPSSMマトリックスで評価した後、ベイズ最適化を設定してDMTサイクルを実行し、最も結合親和性の高いペプチド配列とその結合スコアを見つけ出す過程を追っていきます。
ベイズ最適化(Bayesian Optimization)とは?
ベイズ最適化(BO)は、機械学習の一種で、「評価コストが高い関数(実験やシミュレーション)において、どの入力(今回はペプチド配列)を優先的に評価すれば、最も効率よく最適解にたどり着けるか?」を自律的に判断する手法です。
問題設定
ペプチド配列は20種類のアミノ酸から構成され、9残基のペプチドの場合、理論上は20^9 = 5120億通りもの組み合わせがあります。すべてを実験で評価するのは不可能です。
ベイズ最適化の考え方
「賢く選んで、少ない実験回数で最適解を見つける」手法です。
1. 探索と活用のバランス
- 探索(Exploration): まだ試していない領域を調べる
- 活用(Exploitation): これまでに良い結果が出た領域を詳しく調べる
- この2つをバランスよく行うことで、効率的に最適解に近づきます
2. 予測モデル(ガウス過程)
- 既に評価したペプチドのデータから、未評価のペプチドのスコアを予測します
- 「このペプチドは良いスコアが出そう」という予測値と、「この予測はどれくらい確実か」という不確実性の両方を提供します
3. 獲得関数(次に何を試すか決める)
- 予測値が良いペプチド → 試す価値がある
- 不確実性が高いペプチド → もしかしたら良い結果が出るかもしれない
- この2つを組み合わせて、次に評価すべきペプチドを選びます
具体例で理解する
従来の方法(ランダム探索):
1. ランダムに100個のペプチドを選ぶ
2. 全部評価する
3. 最良のものを選ぶ
→ 運が良ければ良い結果、運が悪ければ無駄が多い
ベイズ最適化:
1. 初期に少数(96個)のペプチドを評価
2. データから「良いペプチドの特徴」を学習
3. 学習結果を基に、次に試すべき96個を賢く選ぶ
4. 評価して、さらに学習を更新
5. これを繰り返す
→ 少ない評価回数で、より良い結果に到達
BOの役割は以下の通りです。
- 初期評価(シードライブラリの生成): まず少数のペプチドを配列ベースの戦略(アラニンスキャニング、ホモログスキャニングなど)で選び、PSSMマトリックスによりその結合親和性(pIC50スコア)を計算(評価)します。
- 代理モデルの構築: 評価結果を基に、未評価のペプチド群のスコア(結合親和性)を予測する機械学習モデル(ガウス過程など)を構築します。
- 獲得関数に基づく選択: 代理モデルの予測結果から、「最もスコアが良さそう(pIC50スコアが低そう)」または「予測の不確実性が高く、評価すればモデルが大きく改善しそう」なペプチドを、獲得関数(Acquisition Function)を使って優先的に選択します。
- 再評価と学習: 選択したペプチドをPSSMマトリックスで実際に評価し、その結果(pIC50スコア)を代理モデルにフィードバック(学習)させることで、モデルの予測精度を向上させます。
このサイクル(Design-Make-Testサイクル)を繰り返すことで、全ペプチドを評価せずとも、最終的に目的とする高性能なペプチド(高い結合親和性を持つペプチド)に効率よくたどり着くことが可能になります。
mobiusは、このBOの仕組みを、ペプチドの設計・評価プロセスに組み込むことを可能にしています。
環境構築
installationの資料はこちら。
注意: mobiusは、ガウス過程モデルやグラフニューラルネットワークなどの機械学習モデルを使用するため、GPUがあると計算が大幅に高速化されます。本記事ではCPUのみを使用した実装を行いますが、大規模な最適化を行う場合はGPUの使用を推奨します。GPUを使用する場合は、Linux環境でCUDA対応のPyTorchをインストールしてください。
Miniforge3のインストール
まず、Miniforge3をインストールします(既にインストール済みの場合はスキップしてください)。
# Miniforge3インストーラをダウンロード
curl -L -O "https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-$(uname)-$(uname -m).sh"
# Miniforge3をインストール
bash Miniforge3-$(uname)-$(uname -m).sh
# インストール後、新しいターミナルを開くか、以下を実行してcondaを有効化# macOS (zsh) の場合eval "$(~/miniforge3/bin/conda shell.zsh hook)"
conda init zsh
source ~/.zshrc
# Linux (bash) の場合eval "$(~/miniforge3/bin/conda shell.bash hook)"
conda init bash
source ~/.bashrc
mobiusのセットアップ
- mobiusリポジトリのクローン
git clone https://git.scicore.unibas.ch/schwede/mobius.git
cd mobius
- 環境ファイルのダウンロード
以下の環境ファイルをダウンロードして、mobiusディレクトリに配置してください。
CPU版(macOS ARM64): environment_cpu.yaml (クローンしたものの中にあるenvironmental.yamlで環境構築もOKですが、私のPCではうまくいかなかったので、修正してます。)
- 仮想環境の作成
# CPU版を使用する場合(macOS ARM64 / Apple Silicon)
mamba env create -f environment_cpu.yaml -n mobius
- 環境のアクティベートとmobiusのインストール
#eval "$(~/miniforge3/bin/mamba shell hook --shell zsh)"
# 環境をアクティベート
mamba activate mobius
注意:
mambaがインストールされていない場合は、conda install mamba -n base -c conda-forge -yでインストールしてください。- 新しいターミナルを開いた場合は、
mamba activate mobiusの前にeval "$(~/miniforge3/bin/mamba shell hook --shell zsh)"(zshの場合)またはeval "$(~/miniforge3/bin/mamba shell hook --shell bash)"(bashの場合)を実行してください。
データの準備
MHCクラスIのPSSMファイルが必要です。mobiusリポジトリには、IEDB(Immune Epitope Database)から取得したPSSMファイルが含まれています。
PSSMファイルについて:
- PSSM(Position-Specific Scoring Matrix:位置特異的スコアリングマトリックス)は、ペプチドの各位置に各アミノ酸が現れる際のスコアを表すマトリックスです。
以下のような構造をしています。
NumCols: 8
A -0.055 -0.024 -0.007 -0.261 0.237 -0.045 -0.050 -0.007
C 0.003 0.085 -0.018 -0.371 0.048 0.005 0.009 -0.020
D -0.006 -0.195 0.029 0.119 -0.494 -0.022 0.017 -0.010
E -0.015 -0.099 0.009 0.036 -0.098 0.008 0.012 -0.005
F 0.011 0.304 -0.064 -0.506 0.018 0.100 0.104 -0.091
G 0.002 0.036 0.009 0.014 -0.912 -0.010 -0.040 -0.007
H 0.023 -0.052 0.015 0.205 -0.566 -0.034 -0.014 -0.028
I -0.004 0.146 -0.056 -0.143 0.312 0.122 0.095 0.098
K 0.018 0.084 0.048 0.233 -0.040 -0.014 -0.040 0.058
L -0.019 0.303 -0.026 -0.218 0.240 0.074 0.057 0.042
M -0.005 0.127 -0.039 0.067 -0.083 0.039 0.049 0.028
N -0.002 -0.148 0.039 0.095 0.075 -0.057 -0.032 -0.007
P -0.015 0.158 0.070 -0.024 0.005 -0.019 -0.139 0.026
Q -0.003 -0.041 0.031 0.128 -0.040 -0.031 -0.061 0.036
R 0.019 0.107 0.042 0.369 0.236 -0.035 -0.084 0.007
S 0.001 -0.597 0.007 0.350 0.333 -0.101 -0.055 0.020
T -0.000 -0.411 0.011 0.170 0.201 -0.053 -0.016 0.015
V -0.006 -0.091 -0.020 -0.085 0.480 0.045 0.045 0.048
W 0.034 0.054 -0.034 -0.130 0.017 0.023 0.057 -0.056
Y 0.020 0.255 -0.047 -0.048 0.032 0.005 0.086 -0.146
Intercept 4.52761
「8つの位置のそれぞれに、どのアミノ酸が好まれるか」を数値化したものです。
行は20種類のアミノ酸、列はペプチドの位置(1〜8)を表します。
各数値は その位置にそのアミノ酸が入ったときの寄与スコア を意味し、
- プラス(+) → 好まれる / 結合に有利
- マイナス(–) → 好まれない / 不利
を示します。
例えば位置5では V(0.480), I(0.312) が高く、
疎水性アミノ酸が強く好まれる位置 と読み取れます。
逆に G(-0.912) は非常に嫌われます。
ペプチド全体のスコアは
各位置のスコアの合計 + 最後の“Intercept(4.52761)”
で計算され、MHC結合予測や免疫原性予測に使われます。
PSSMファイルはmobius/data/IEDB_MHC_I-2.9_matx_smm_smmpmbec/smmpmbec_matrix/ディレクトリに配置されています。
クローンしてきたmobiusのexamplesのhello_world.ipynbを参考に、mobius_test/linear_peptide/linear_peptide.ipynbを作成して実行してください。
cursorでのjupyter環境の設定は以下をご覧ください。
📰 環境構築|【完全版】In silico創薬実践書 〜おうち創薬で論文を書こう〜
mobiusによるベイズ最適化の実行
全コードはこちらを参考にしています。チュートリアルとほぼ一緒ですが、一部説明を追加しています。
# ----------------------------------------------------
# STEP 1: 準備と環境設定
# ----------------------------------------------------
# 必要なライブラリをインポート
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from gpytorch.priors import NormalPrior
# mobiusの主要なクラスと関数をインポート
from mobius import Planner, SequenceGA # プランナーと遺伝的アルゴリズム最適化器
from mobius import Map4Fingerprint # 分子フィンガープリント(MAP4)
from mobius import GPModel, ExpectedImprovement, TanimotoSimilarityKernel # ガウス過程モデル、獲得関数、カーネル
from mobius import LinearPeptideEmulator # MHCクラスIエミュレーター(PSSMベース)
from mobius import homolog_scanning, alanine_scanning # 配列ベースのスキャニング戦略
from mobius import convert_FASTA_to_HELM # FASTA形式からHELM形式への変換
# ----------------------------------------------------
# STEP 2: LinearPeptideEmulatorの初期化
# ----------------------------------------------------
# MHCクラスI HLA-A*02:01のLinearPeptideEmulatorを初期化
# Position Specific Scoring Matrix (PSSM) ファイルを使用
import os
# PSSMファイルのパス(複数のペプチド長に対応)
pssm_dir = os.path.join('..', '..', 'mobius', 'data', 'IEDB_MHC_I-2.9_matx_smm_smmpmbec', 'smmpmbec_matrix')
pssm_dir = os.path.normpath(os.path.abspath(pssm_dir))
pssm_files = [
os.path.join(pssm_dir, 'HLA-A-02:01-8.txt'),
os.path.join(pssm_dir, 'HLA-A-02:01-9.txt'),
os.path.join(pssm_dir, 'HLA-A-02:01-10.txt'),
os.path.join(pssm_dir, 'HLA-A-02:01-11.txt')
]
# LinearPeptideEmulatorを初期化
# 実際の使用では、このステップを実際のラボ実験に置き換える必要があります
lpe = LinearPeptideEmulator(pssm_files)
# ----------------------------------------------------
# STEP 3: リードペプチドの定義
# ----------------------------------------------------
# 最適化の出発点となるリードペプチドを定義
# FASTA形式('HMTEVVRRC')からHELM形式に変換
# mobiusは内部的にHELM記法を使用してペプチド配列をエンコードします
lead_peptide = convert_FASTA_to_HELM('HMTEVVRRC')[0]
print(f"リードペプチド(HELM形式): {lead_peptide}")
# ----------------------------------------------------
# STEP 4: シードライブラリの生成
# ----------------------------------------------------
# 初期評価用のシードライブラリを生成
# アラニンスキャニングとホモログスキャニングを組み合わせて96個のペプチドを生成
seed_library = [lead_peptide]
# アラニンスキャニング: 各位置をアラニンに置き換えた変異体を生成
for seq in alanine_scanning(lead_peptide):
seed_library.append(seq)
# ホモログスキャニング: 各位置を類似アミノ酸に置き換えた変異体を生成
for seq in homolog_scanning(lead_peptide):
seed_library.append(seq)
if len(seed_library) >= 96:
print('Reach max. number of peptides allowed.')
break
# ----------------------------------------------------
# STEP 5: シードライブラリの評価
# ----------------------------------------------------
# シードライブラリの各ペプチドをLinearPeptideEmulatorで評価
# HELM形式のペプチドを評価し、pIC50スコアを計算
pic50_seed_library = lpe.score(seed_library)
print(f"評価したペプチド数: {len(seed_library)}")
print(f"平均pIC50スコア: {np.mean(pic50_seed_library):.3f}")
print(f"最良のスコア: {np.min(pic50_seed_library):.3f}")
best_idx = np.argmin(pic50_seed_library)
print(f"最良のペプチド: {seed_library[best_idx]}")
print(f"(スコアが低いほど結合親和性が高い)")
# ----------------------------------------------------
# STEP 6: ベイズ最適化の設定
# ----------------------------------------------------
# 1. 分子フィンガープリント(MAP4)を設定
# HELM形式の入力を受け取り、4096次元のベクトルに変換
# 構造的に類似したペプチドが近いベクトル表現になる
map4 = Map4Fingerprint(input_type='helm', dimensions=4096, radius=1)
# 2. ガウス過程モデルを設定
# Tanimoto類似度カーネルを使用し、MAP4フィンガープリントで変換
# 既に評価したペプチドのデータから、未評価のペプチドのスコアを予測
gpmodel = GPModel(kernel=TanimotoSimilarityKernel(), transform=map4, noise_prior=NormalPrior(loc=0, scale=1))
# 3. 獲得関数(Expected Improvement)を設定
# ガウス過程モデルを使用し、最小化問題(pIC50スコアを小さくする)として設定
# pIC50スコアは低いほど結合親和性が高いため、maximize=False
# 次に評価すべきペプチドを選択するための関数
acq = ExpectedImprovement(gpmodel, maximize=False)
# ----------------------------------------------------
# STEP 7: デザインプロトコルの定義
# ----------------------------------------------------
# YAML形式でデザインプロトコルを定義
# ペプチドの構造制約とフィルター条件を指定
yaml_content = """
design:
monomers:
default: [A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y]
APOLAR: [A, F, G, I, L, P, V, W]
POLAR: [C, D, E, H, K, N, Q, R, K, S, T, M]
AROMATIC: [F, H, W, Y]
POS_CHARGED: [K, R]
NEG_CHARGED: [D, E]
polymers:
- PEPTIDE1{X.M.X.X.X.X.X.X.X}$$$$V2.0:
PEPTIDE1:
1: [AROMATIC, NEG_CHARGED]
4: POLAR
9: [A, V, I, L, M, T]
filters:
- class_path: mobius.PeptideSelfAggregationFilter
- class_path: mobius.PeptideSolubilityFilter
init_args:
hydrophobe_ratio: 0.5
charged_per_amino_acids: 5
"""
# デザインプロトコルをYAMLファイルに保存
with open('design_protocol.yaml', 'w') as f:
f.write(yaml_content)
# ----------------------------------------------------
# STEP 8: 最適化器とプランナーの初期化
# ----------------------------------------------------
# 遺伝的アルゴリズム(GA)ベースの最適化器を設定
# period=15: GAの世代数を15に設定
optimizer = SequenceGA(algorithm='GA', period=15, design_protocol_filename='design_protocol.yaml')
# プランナーを初期化(獲得関数と最適化器を組み合わせる)
ps = Planner(acq, optimizer)
# ----------------------------------------------------
# STEP 9: Design-Make-Test(DMT)サイクルの実行
# ----------------------------------------------------
# 評価済みペプチドとスコアを保持するリスト
peptides = seed_library.copy()
pic50_scores = pic50_seed_library.copy()
# 後で分析するためのデータを保存
data = [(0, p, s) for p, s in zip(peptides, pic50_scores)]
# 3回のDMTサイクルを実行
num_cycles = 3
batch_size = 96
for i in range(num_cycles):
print(f'\n=== DMTサイクル {i+1}/{num_cycles} ===')
# Design: ベイズ最適化に基づいて、次に評価すべきペプチドを推薦
suggested_peptides, _ = ps.recommend(peptides, pic50_scores.reshape(-1, 1), batch_size=batch_size)
# Test: 推薦されたペプチドをLinearPeptideEmulatorで評価
# 注意: 実際の使用では、ここで実際のラボ実験を実行します
pic50_suggested_peptides = lpe.score(suggested_peptides)
# 新しいデータを既存のリストに追加
peptides = np.concatenate([peptides, suggested_peptides])
pic50_scores = np.concatenate((pic50_scores, pic50_suggested_peptides), axis=0)
data.extend([(i + 1, p, s) for p, s in zip(suggested_peptides, pic50_suggested_peptides)])
# これまでに見つかった最良のペプチドとそのスコアを表示
best_seq = peptides[np.argmin(pic50_scores)]
best_pic50 = np.min(pic50_scores)
print(f'\n最良のペプチド: {best_seq}')
print(f'最良のスコア: {best_pic50:.3f}')
print(f'(スコアが低いほど結合親和性が高い)')
# データをDataFrameに変換(可視化用)
df = pd.DataFrame(data=data, columns=('iter', 'polymer', 'exp_value'))
# ----------------------------------------------------
# STEP 10: 結果の可視化
# ----------------------------------------------------
# 最適化の進行状況を可視化
fig, ax = plt.subplots(figsize=(20, 7.5))
# イテレーション番号を文字列に変換('Init.'は初期シードライブラリ)
df['iter'] = df['iter'].replace({0: 'Init.', 1: '1', 2: '2', 3: '3'})
# 各イテレーションでのpIC50スコアの平均と95%信頼区間をプロット
sns.lineplot(x='iter', y='exp_value', data=df, ax=ax,
errorbar=('ci', 95), err_style='bars',
linewidth=3.5, err_kws={'lw': 3.5})
# グラフの設定
ax.set_xlim([-0.9, 3.5])
ax.set_ylim([-2, 4.5])
ax.set_ylabel('pIC50', fontsize=30)
ax.set_xlabel('Generations', fontsize=30)
ax.xaxis.set_tick_params(labelsize=30)
ax.yaxis.set_tick_params(labelsize=30)
# グラフのスタイル調整
sns.despine()
plt.tight_layout()
plt.show()
# 最終結果の表示
print('\n' + '='*60)
print('最適化結果のサマリー')
print('='*60)
best_idx = np.argmin(pic50_scores)
best_peptide = peptides[best_idx]
best_score = pic50_scores[best_idx]
print(f'\n最良のペプチド配列(HELM形式):')
print(f' {best_peptide}')
print(f'\npIC50スコア: {best_score:.3f}')
print(f'(スコアが低いほど結合親和性が高い)')
print(f'\n評価したペプチドの総数: {len(peptides)}')
print(f'初期シードライブラリ: {len(seed_library)}')
print(f'DMTサイクルで評価したペプチド: {len(peptides) - len(seed_library)}')
print('='*60)
全体の流れ:AIを使った新しいペプチドの探索
このコードは、既存のリードペプチドをベースに、AI(ベイズ最適化)を使って、ターゲットタンパク質(MHCクラスI)に最もよく結合するであろう新しいペプチド配列を効率的に見つけ出すという、創薬研究のプロセスをシミュレーションしています。
プロセスは以下のステップで進行します。
- 準備と初期設定(Step 1〜3):
- 必要なツールを準備し、MHCクラスIのPSSMマトリックスを読み込んでLinearPeptideEmulatorを初期化します。
- 最適化の出発点となるリードペプチドを定義し、HELM形式に変換します。
- シードライブラリの生成と評価(Step 4〜5):
- リードペプチドからアラニンスキャニングとホモログスキャニングを用いて96個の変異体を生成します。
- 生成したペプチドをPSSMマトリックスで評価し、pIC50スコア(結合スコア)を計算します。これはAIが学習するための最初のデータセットとなります。
- ベイズ最適化の設定(Step 6〜8):
- 分子フィンガープリント(MAP4)、ガウス過程モデル、**獲得関数(Expected Improvement)**を設定します。
- デザインプロトコル(YAML形式)で、ペプチドの構造制約とフィルター条件を定義します。
- 最適化器(遺伝的アルゴリズム)とプランナーを初期化します。
- Design-Make-Testサイクル(Step 9):
- AIが学習結果に基づき、「次に計算すれば最も良いスコアが出そう」と予測した最適な96個のペプチドを選び出し、再度PSSMマトリックスで評価します。
- このサイクルを3回繰り返します。
- 各サイクル終了時に、これまでに見つかった最良のペプチドとその結合スコアを表示します。
- 結果の可視化(Step 10):
- 各イテレーションでのpIC50スコアの分布を可視化し、最適化の進行状況を確認します。
- 最終的に、最良のペプチド配列とその結合スコアを表示します。
STEP 1: 準備と環境設定
シミュレーションを行うための道具の準備を行います。
import numpy as np/import pandas as pd/import matplotlib.pyplot as plt/import seaborn as sns: 数値計算のNumPy、データ分析のPandas、可視化のMatplotlibとSeabornという標準的なPythonライブラリをプログラムに組み込んでいます。from mobius import Planner, SequenceGA:mobiusから、ベイズ最適化のプランナーと、遺伝的アルゴリズムベースの最適化器を呼び出せるようにしています。from mobius import Map4Fingerprint: ペプチド配列を数値ベクトル(フィンガープリント)に変換するMAP4という手法を呼び出しています。from mobius import GPModel, ExpectedImprovement, TanimotoSimilarityKernel: ベイズ最適化の核心となるガウス過程モデル、獲得関数、カーネル関数を呼び出しています。from mobius import LinearPeptideEmulator: PSSMマトリックスを使用してペプチドの結合親和性を予測するLinearPeptideEmulatorを呼び出しています。from mobius.utils import MolFromHELM: HELM形式のペプチドをRDKit分子に変換する関数を呼び出しています。from mobius import homolog_scanning, alanine_scanning: 初期ライブラリを生成するための配列ベースのスキャニング戦略を呼び出しています。from mobius import convert_FASTA_to_HELM: FASTA形式のペプチド配列を、mobiusが使用するHELM形式に変換する関数を呼び出しています。
STEP 2: LinearPeptideEmulatorの初期化
MHCクラスI(HLA-A*02:01)のPSSMファイルを読み込み、LinearPeptideEmulatorを初期化します。
IEDBデータベースについて
PSSMファイルは、IEDB(Immune Epitope Database)から取得された実験データに基づいて作成されています。
データの取得方法:
- 文献からの収集: 公開された科学論文から手作業で実験データを収集
- 実験データ: 実際のMHC結合アッセイで測定された結合親和性(IC50値など)
- 機械学習モデル: SMM(Stabilized Matrix Method)という機械学習モデルで学習
- PSSMファイル生成: 学習したモデルからPSSMファイルが生成される
pssm_files = [...]: 複数のペプチド長(8、9、10、11残基)に対応するPSSMファイルのパスをリストで指定しています。lpe = LinearPeptideEmulator(pssm_files): LinearPeptideEmulatorを初期化しています。これにより、ペプチドをMHCクラスIに結合させた際のpIC50スコア(結合スコア)を計算できます。pIC50スコアは低いほど結合親和性が高いことを示します。
STEP 3: リードペプチドの定義
最適化の出発点となるリードペプチドを定義します。
lead_peptide = convert_FASTA_to_HELM('HMTEVVRRC')[0]: FASTA形式のペプチド配列('HMTEVVRRC')を、mobiusが使用するHELM形式に変換しています。HELM(Hierarchical Editing Language for Macromolecules)は、ペプチドやその他の複雑な生体分子を表現するための標準形式です。
HELM形式の表記について
このノートブックでは、ペプチド配列をHELM形式(Hierarchical Editing Language for Macromolecules)で表現しています。
HELM形式の構造:
例: PEPTIDE1{H.M.M.D.F.I.F.E.V}$$$$V2.0
この表記は以下の部分から構成されています:
PEPTIDE1: ペプチドのタイプ- 線形ペプチドを表す識別子
{H.M.M.D.F.I.F.E.V}: アミノ酸配列- 各アミノ酸は1文字コードで表される
- ドット(
.)で区切られている - 左から右へ、N末端からC末端の順
$$$$: セパレーター- 複数のポリマーや接続情報を区切る
- 線形ペプチドの場合は接続情報がないため、
$$$$のみ
V2.0: HELMのバージョン- HELM記法のバージョン番号
STEP 4: シードライブラリの生成
初期評価用のシードライブラリを生成します。
seed_library = [lead_peptide]: シードライブラリのリストを初期化し、リードペプチドを最初の要素として追加しています。for seq in alanine_scanning(lead_peptide):: アラニンスキャニングを実行します。これは、リードペプチドの各位置をアラニン(A)に置き換えた変異体を生成する戦略です。各位置の重要性を評価するために使用されます。for seq in homolog_scanning(lead_peptide):: ホモログスキャニングを実行します。これは、リードペプチドの各位置を類似アミノ酸に置き換えた変異体を生成する戦略です。構造的に類似したアミノ酸への置換により、機能を維持しながら多様性を導入します。if len(seed_library) >= 96: break: シードライブラリが96個に達したら、ループを終了します。
STEP 5: シードライブラリの評価
生成したシードライブラリの各ペプチドをPSSMマトリックスで評価します。
pic50_seed_library = lpe.score(seed_library): LinearPeptideEmulatorを使用して、シードライブラリの各ペプチドのpIC50スコアを計算しています。pIC50スコアは低いほど結合親和性が高いことを示します。
このステップでは、96個のペプチドそれぞれに対してPSSMマトリックスによる評価を実行し、MHCクラスIとの結合スコアを取得します。これらのスコアは、ベイズ最適化の学習データとして使用されます。
STEP 6: ベイズ最適化の設定
ベイズ最適化に必要なコンポーネントを設定します。
map4 = Map4Fingerprint(input_type='helm', dimensions=4096, radius=1): MAP4フィンガープリントを設定しています。これは、HELM形式のペプチド配列を4096次元の数値ベクトルに変換する手法です。radius=1は、原子から1ホップ以内の部分構造を考慮することを意味します。
役割: ペプチド配列を数値ベクトルに変換する「翻訳機」
なぜ必要?
- コンピュータは文字列(ペプチド配列)を直接理解できません
- 数値ベクトルに変換することで、数学的な計算が可能になります
具体例:
ペプチド「HMTEVVRRC」→ [0.23, -0.15, 0.87, ..., 0.42] (4096次元のベクトル)
ペプチド「HMTEVVRRV」→ [0.24, -0.14, 0.86, ..., 0.41] (似た配列なので似たベクトル)
gpmodel = GPModel(kernel=TanimotoSimilarityKernel(), transform=map4, noise_prior=NormalPrior(loc=0, scale=1)): ガウス過程モデルを設定しています。これは、ペプチドのフィンガープリントから結合親和性を予測する代理モデルです。TanimotoSimilarityKernelは、2つのペプチドの類似度を計算するカーネル関数です。noise_priorは、観測ノイズの事前分布を設定しています。
役割: 「予測屋さん」- 未評価のペプチドのスコアを予測する
どのように予測するか?
-
学習: 既に評価したペプチドのデータを見る
評価済み: HMTEVVRRC → スコア 2.5 評価済み: HMTEVVRRV → スコア 2.3 評価済み: HMTEVVRRA → スコア 2.8 -
パターン発見: 「似た配列は似たスコアになる」というパターンを学習
「位置9がC, V, Aのとき、スコアは2.3-2.8の範囲」 「位置8がRのとき、スコアが良くなる傾向」 -
予測: 未評価のペプチドのスコアを予測
未評価: HMTEVVRRL → 予測スコア: 2.4 ± 0.3 (「2.4くらいになりそう。でも、0.3の誤差があるかも」)
acq = ExpectedImprovement(gpmodel, maximize=False): 獲得関数(Expected Improvement)を設定しています。これは、次に評価すべきペプチドを選ぶための関数です。maximize=Falseは、pIC50スコアを最小化する(結合親和性を最大化する)問題であることを示しています。pIC50スコアは低いほど良いため、最小化問題として設定します。
役割: 「次に何を試すか決める審判員」
判断基準:
-
予測値が良いペプチド → 試す価値がある
予測スコア: 1.5(現在の最良: 2.0より良い!) → 試してみたい -
不確実性が高いペプチド → もしかしたら良い結果が出るかも
予測スコア: 2.5 ± 1.0(不確実性が大きい) → もしかしたら1.5になるかもしれない! -
バランス: この2つを組み合わせて、期待改善値を計算
期待改善値 = 「このペプチドを試すことで、どれくらい改善が期待できるか」
STEP 7: デザインプロトコルの定義
ペプチドの構造制約とフィルター条件をYAML形式で定義します。
yaml_content = """...""": YAML形式の文字列でデザインプロトコルを定義しています。monomers: 各位置で使用可能なアミノ酸のセットを定義しています。defaultは全20種類の標準アミノ酸、APOLARは非極性アミノ酸、POLARは極性アミノ酸など、意味のあるグループに分類されています。polymers: ペプチドのスキャフォールドを定義しています。PEPTIDE1{X.M.X.X.X.X.X.X.X}$$$$V2.0は、9残基の線形ペプチドを表しています。各位置(1, 4, 9)で使用可能なアミノ酸のセットを指定しています。filters: 合成や溶解度に問題がある可能性のあるペプチドを除外するフィルターを定義しています。PeptideSelfAggregationFilterは自己凝集を防ぐフィルター、PeptideSolubilityFilterは溶解度を確保するフィルターです。
with open('design_protocol.yaml', 'w') as f: f.write(yaml_content): 定義したデザインプロトコルをYAMLファイルに保存しています。
STEP 8: 最適化器とプランナーの初期化
ベイズ最適化を実行するための最適化器とプランナーを初期化します。
optimizer = SequenceGA(algorithm='GA', period=15, design_protocol_filename='design_protocol.yaml'): 遺伝的アルゴリズム(GA)ベースの最適化器を設定しています。algorithm='GA'は遺伝的アルゴリズムを使用することを指定し、period=15はGAの世代数を15に設定しています。design_protocol_filenameで、先ほど定義したデザインプロトコルファイルを指定しています。ps = Planner(acq, optimizer): プランナーを初期化しています。プランナーは、獲得関数(acq)と最適化器(optimizer)を組み合わせて、次に評価すべきペプチドを推薦する役割を担います。
STEP 9: Design-Make-Testサイクルの実行
ベイズ最適化のメインループを実行します。
peptides = seed_library.copy()/pic50_scores = pic50_seed_library.copy(): 評価済みのペプチドとそのpIC50スコアを保持するリストを初期化しています。data = [(0, p, s) for p, s in zip(peptides, pic50_scores)]: 後で可視化するためのデータを保存しています。0は初期シードライブラリを表します。for i in range(3):: 3回のDMTサイクルを実行します。suggested_peptides, _ = ps.recommend(peptides, pic50_scores.reshape(-1, 1), batch_size=96): プランナーが、既存のデータ(peptidesとpic50_scores)に基づいて、次に評価すべき96個のペプチドを推薦します。ベイズ最適化のアルゴリズムが、獲得関数と最適化器を使って最適なペプチドを選び出します。pic50_suggested_peptides = lpe.score(suggested_peptides): 推薦された各ペプチドをPSSMマトリックスで評価し、pIC50スコアを計算します。peptides = np.concatenate([peptides, suggested_peptides])/pic50_scores = np.concatenate(...): 新しいデータを既存のリストに追加します。best_idx = np.argmin(pic50_scores): これまでに見つかった最良のペプチドのインデックスを取得します(スコアが最小のもの)。best_seq = peptides[best_idx]/best_pic50 = pic50_scores[best_idx]: 最良のペプチド配列とその結合スコアを表示します。
DMTサイクルの流れ(詳しく)
1. Design(設計)フェーズ
何をしているか?
- ガウス過程モデルが、これまでに評価した全てのペプチドのデータを分析
- 「どのような特徴を持つペプチドが良いスコアを出すか」を学習
- 獲得関数が、この学習結果を基に「次に試すべき96個のペプチド」を選ぶ
具体例:
学習したパターン:
- 位置1にH(ヒスチジン)があるとスコアが良い
- 位置4にE(グルタミン酸)があるとスコアが良い
- 位置9にV(バリン)があるとスコアが良い
→ これらの特徴を組み合わせたペプチドを推薦
2. Make(生成)フェーズ
何をしているか?
- 推薦されたペプチド配列を準備
- デザインプロトコルに基づいて、化学的に妥当なペプチドのみを生成
- フィルターで、実用的でないペプチドを除外
注意: このノートブックでは、ペプチド配列は既に生成されているため、このステップは自動的に行われます。
3. Test(評価)フェーズ
何をしているか?
- 推薦された96個のペプチドを、LinearPeptideEmulatorで評価
- 各ペプチドの結合スコア(pIC50)を取得
実際の使用では:
- ここで実際のラボ実験を実行します
- 分子ドッキング、結合アッセイなど
4. 更新フェーズ
何をしているか?
- 新しい96個のデータを、既存のデータに追加
- ガウス過程モデルを更新(新しいデータで再学習)
- これまでに見つかった最良のペプチドを記録
学習の進化:
サイクル1: 96個のデータで学習 → 次の96個を選ぶ
サイクル2: 192個のデータで学習(より正確に!) → 次の96個を選ぶ
サイクル3: 288個のデータで学習(さらに正確に!) → 次の96個を選ぶ
STEP 10: 結果の可視化
最適化の進行状況を可視化します。
df = pd.DataFrame(data=data, columns=('iter', 'polymer', 'exp_value')): 保存したデータをPandasのDataFrameに変換します。df['iter'] = df['iter'].replace({0: 'Init.', 1: '1', 2: '2', 3: '3'}): イテレーション番号を文字列に変換します('Init.'は初期シードライブラリを表します)。sns.lineplot(...): 各イテレーションでのpIC50スコアの平均と95%信頼区間を可視化します。これにより、最適化が進行するにつれてpIC50スコアが改善(減少)するかを確認できます。- 最終結果の表示: 最適化の完了後、最良のペプチド配列(HELM形式)とその結合スコア(pIC50スコア)を表示します。また、評価したペプチドの総数も表示されます。
結果の可視化
実行が完了すると、各イテレーションでのpIC50スコアの分布が可視化されます。グラフから、最適化が進行するにつれてpIC50スコアが改善(減少)していく様子が確認できます。
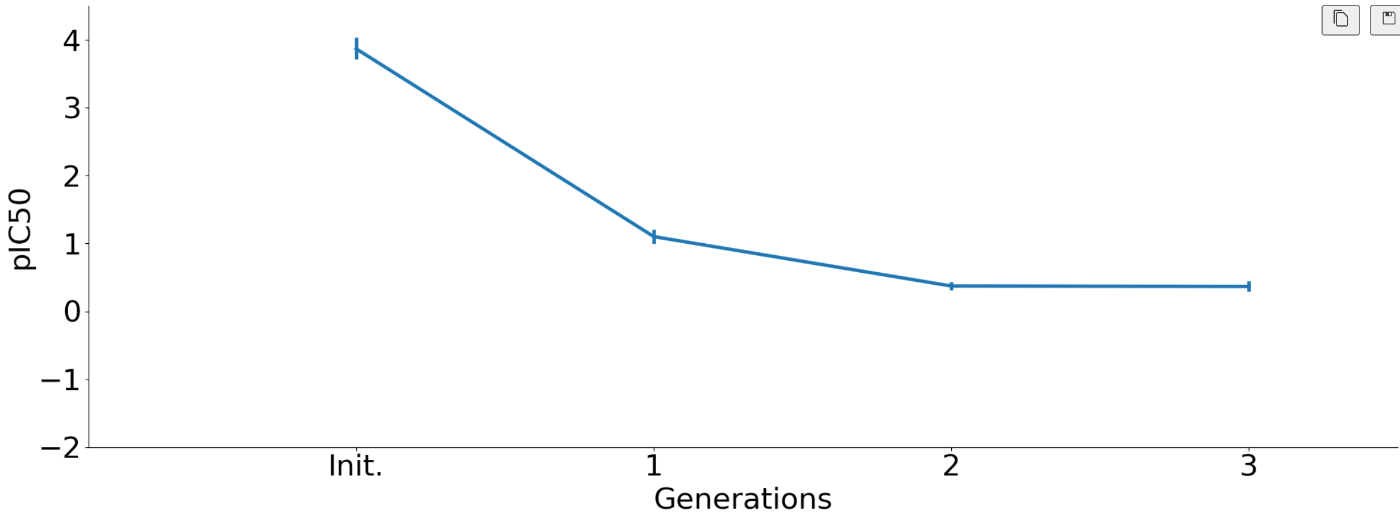
また、各イテレーションの終了時に、これまでに見つかった最良のペプチドとそのスコアが表示されます。最終結果として、以下のような出力が得られます。
============================================================
最適化結果のサマリー
============================================================
最良のペプチド配列(HELM形式):
PEPTIDE1{F.M.M.E.D.I.F.R.V}$$$$V2.0
pIC50スコア: -0.709
(スコアが低いほど結合親和性が高い)
評価したペプチドの総数: 384
初期シードライブラリ: 96
DMTサイクルで評価したペプチド: 288
============================================================
これは、HELM形式で表現された最良のペプチド配列と、そのpIC50スコア(-0.335)を示しています。pIC50スコアは低いほど結合親和性が高く、初期のリードペプチドから大幅に改善されていることがわかります。
最後に
mobiusとベイズ最適化を活用したペプチド最適化、いかがでしたでしょうか。
数千にも及ぶ大規模な仮想ペプチドライブラリの中から、わずか数回のDMTサイクルで最も結合親和性が高いと予測されるペプチド配列を効率的に見つけ出すことができました。
全ての仮想ライブラリを網羅的に計算することは、膨大な実験リソースと時間が必要となり現実的ではありません。しかし、ベイズ最適化は初期の限られたデータからAIが学習し、次に試すべき最適なペプチドを予測することで、リソースを大幅に節約しながら、有望なペプチドに素早くたどり着くための強力な探索手法となります。
ぜひ皆さんもmobiusとベイズ最適化を組み合わせ、効率的なインシリコ創薬に取り組んでみてください!
参考文献
- mobius Documentation
- mobius GitHub Repository
- Eberhardt, J., Lees, A., Lill, M., & Schwede, T. (2024). Combining Bayesian optimization with sequence- or structure-based strategies for optimization of protein-peptide binding. ChemRxiv. https://doi.org/10.26434/chemrxiv-2023-b7l81-v2
License: Apache-2.0
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

