

この記事は機械学習と強化学習を使用して、さらに結合力の高い環状ペプチドを発見するin silicoペプチドスクリーニングの記事です。
すでに結合がわかっている環状ペプチド(既存もしくはRF PeptideとProteinMPNNから判明)をベースにさらに結合力の高い環状ペプチドを予測することができます。
最先端の機械学習モデル、強化学習モデルを組み合わせた手法になっているので、ぜひ参考にしてみてください!
【この記事のまとめ】
創薬研究者やバイオインフォマティクスに関心がある方に向けて、機械学習(ML)による活性予測と強化学習(RL)を組み合わせ、膨大な探索空間から「高活性な環状ペプチド」を自律的に効率よく特定する手法を解説します。
- 強化学習による知的探索の自動化:単なるランダムスクリーニングではなく、強化学習エージェントが「より高い報酬(活性)」を求めて配列を最適化する高度な探索プロセスを学べます。
- ML代理モデルによる高速評価:ドッキングシミュレーション等の重い計算を機械学習モデルで近似(代替)し、計算コストを抑えつつ膨大な数の配列をスクリーニングするパイプラインを構築します。
- Pythonによる実践的ワークフロー:環境構築から報酬設計(Reward Design)、学習曲線の可視化まで、実際のコードを交えてハンズオン形式で紹介しています。
この記事を読むことで、AIが自ら「最適な配列」を学習・提案する次世代のスクリーニング環境を、自身のPCやクラウド環境に構築できるようになります。
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700,
CPU: Corei7-13700F, メモリ:32GB
GPU: GeForce RTX 4070 VENTUS 2X 12G OC

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


分子ドッキングを使ったIn silico screening
インシリコスクリーニングとは、コンピューター上で多数の化合物を扱い、標的となるタンパク質などに結合しそうな分子を効率よく探し出す手法です。実験を行う前段階として用いられ、創薬研究における探索工程を大幅に高速化できる点が特徴です。
その代表的な方法の一つが分子ドッキングを用いたインシリコスクリーニングです。分子ドッキングでは、化合物が標的分子のどこに、どのような姿勢で結合するかを計算機上で予測し、その結合の強さをスコアとして評価します。
このスコアを指標にすることで、数万〜数百万といった膨大な化合物の中から、標的に対して結合力が高いと予測される候補分子を効率的に絞り込むことが可能になります。分子ドッキングを用いたインシリコスクリーニングは、「まず当たりをつける」ための手法として、現在の創薬研究において欠かせないアプローチとなっています。
分子ドッキングを用いたインシリコスクリーニングは強力な手法ですが、一方で課題も存在します。特に、スクリーニングに用いる化合物がランダムに生成されたものである場合、膨大な候補の中から高い結合力を持つ化合物に到達するまでに多くの計算資源と時間を要します。つまり、「当たり」を引くまでの効率が必ずしも高くないという問題があります。
機械学習、強化学習を使ったIn silico screening
この課題に対して有効なのが、機械学習や強化学習を用いたインシリコスクリーニングです。これらの手法では、過去の評価結果(例えば分子ドッキングスコア)をもとに学習を行い、「次に高い結合力を示しそうな化合物」や「有望な配列」を意図的に提案することができます。
さらに、機械学習や強化学習によって提案された化合物を分子ドッキングで評価することで、計算機上での予測と実測(ドッキング評価)を組み合わせた効率的な探索が可能になります。
これまで環状ペプチドの機械学習・強化学習には Mobiusというパッケージを紹介してきましたが、実運用の面では扱いづらい点もありました。そこで本記事では、環状ペプチドのインシリコスクリーニングをより直感的かつ柔軟に行えるよう、新たに一から開発した手法を紹介します。
この手法を用いることで、読者の皆さんが興味を持つ環状ペプチドと標的分子の組み合わせに対して、機械学習・強化学習と分子ドッキングを組み合わせたスクリーニングを実践し、結合力の高い候補分子を効率よく見つけることが可能になります。
環境構築
仮想環境の構築
こちらに開発したコードを記載しています。
以下を行い、In_silico_screening環境をセットアップしてみてください。
#githubレポジトリのダウンロード
git clone https://github.com/Barashin/In_silico_screening_of_macrocyclic_peptides.git
#In_silico_screening_of_macrocyclic_peptidesフォルダへ
cd /home/shizuku/In_silico_screening_of_macrocyclic_peptides
#GNN + Active Learning環境の設定
bash setup_in_silico_screening.sh
# AutoDock CrankPep環境の設定
bash adcpsuite_micromamba.shまた以下でテストを行なってください。
#環境のアクティベート
micromamba activate in_silico_screening
#環境テスト
python test_environment.pyインプットファイルの設定
以下のディレクトリ構造を参考に
Inputフォルダ:標的タンパク質(例:1O6K_noligand.pdb)、結合ペプチド(例:Peptideligand.pdbqt)
docking_setupフォルダ:結合場所の内容が入ったファイル(例:1O6K.trg)
を作成してください。
Research_Linux/
├── Research/ # Pythonモジュール(必須)
│ ├── active_learning_gnn.py
│ ├── transformer_models.py
│ └── adcp_interface.py
│
├── Input/ # 入力ファイル(.trg作成時に必要)
│ ├── 1O6K_noligand.pdb
│ └── Peptideligand.pdbqt
│
├── docking_setup/ # ターゲットファイル(必須)
│ └── 1O6K.trg
│
├── result/ # 初期ドッキング結果(自動作成)
├── result_active_learning/ # AL結果(自動作成)
└── al_output/ # 最終出力(自動作成)全実装概要
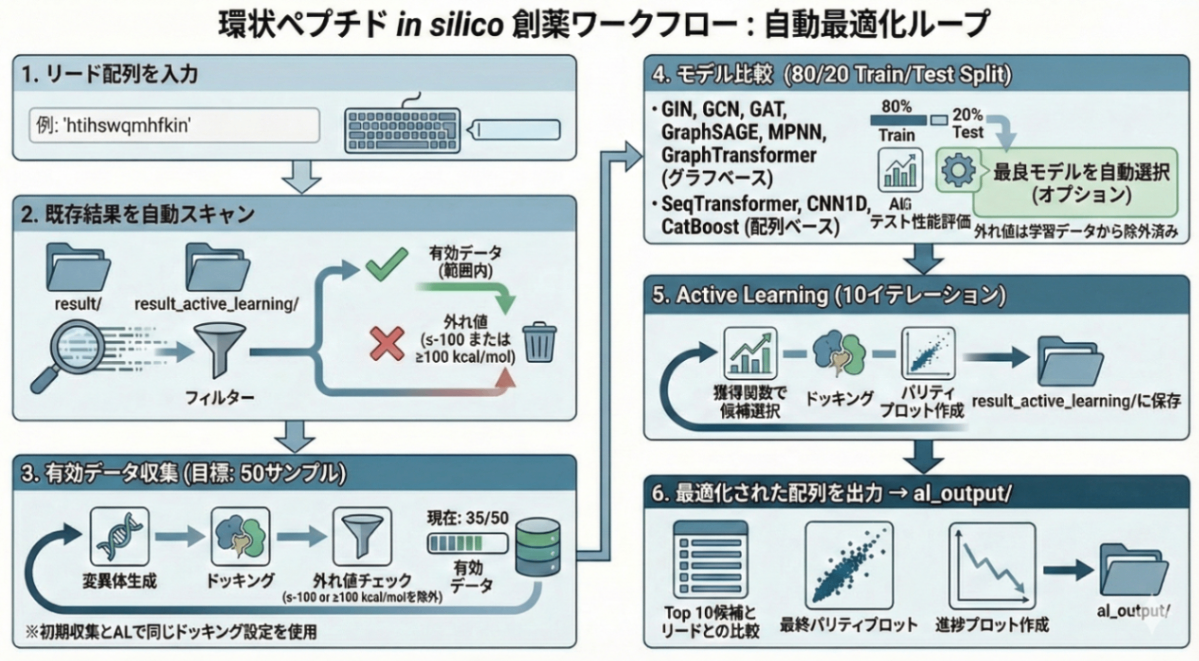
1. リード入力: 最適化の起点となる親配列を決定する。
2. データ洗浄: 既存ファイルを走査し、外れ値を自動除外する。
3. 初期収集: 有効データが50件(変更可能)に達するまで、自動変異とドッキング(Autodock CrankPep)を繰り返す。
4. モデル選定: 9種類のアルゴリズムを比較し、最も予測精度の高いモデルを採用する。
5. 能動学習: AIが選んだ有望候補をドッキングし、10回ループで精度と活性を磨き上げる。
6. 結果出力: 改良されたTop 10配列と、進捗を可視化した最終レポートを生成する。
コードの実行
コードは以下で実行できる
python active_learning_from_lead.py --lead "htihswqmhfkin"デフォルト設定だと、50個の環状ペプチドを—lead に記入した配列から自動生成し、その後の分子ドッキング→機械学習→強化学習へと進む
以下のような引数で、いろいろなパラメータを設定できる。
| 引数 | 短縮 | デフォルト | 説明 |
|------|------|-----------|------|
| `--lead` | `-l` | (必須) | リード配列 |
| `--existing-dir` | | result/ | 既存結果のディレクトリ |
| `--iterations` | `-i` | 10 | ALイテレーション数 |
| `--batch-size` | `-b` | 5 | 各イテレーションの配列数 |
| `--runs` | `-N` | 5 | MC探索回数 |
| `--evals` | `-e` | 100000 | 評価ステップ数 |
| `--timeout` | | 600 | ドッキングタイムアウト(秒) |
| `--acquisition` | | EI | 獲得関数 (EI/UCB/PI) |
| `--min-usable-samples` | | 50 | 学習に必要な最小サンプル数(外れ値除く) |
| `--auto-select-model` | | False | 最良モデルを自動選択 |
| `--production` | | | 本番設定 (timeout=1200) |
| `--quick` | | | クイックテスト (N=1, n=5000, min_usable=20) |
| `--no-confirm` | | | 確認スキップ |結果
ペプチドライブラリの生成
まずleadの配列をランダムに変異させた50個(デフォルト値変更可能)の配列が生成する
モデルの比較
次にその配列と結合力を使用し、様々な機械学習モデルが作成され、比較される。
評価指標(R2)を用いて、Bestなものが自動的に選ばれるが、ぜひ目で見て確認してほしい。(CatBoostが過学習気味ではあるが、結構良さそうな感じでもある)
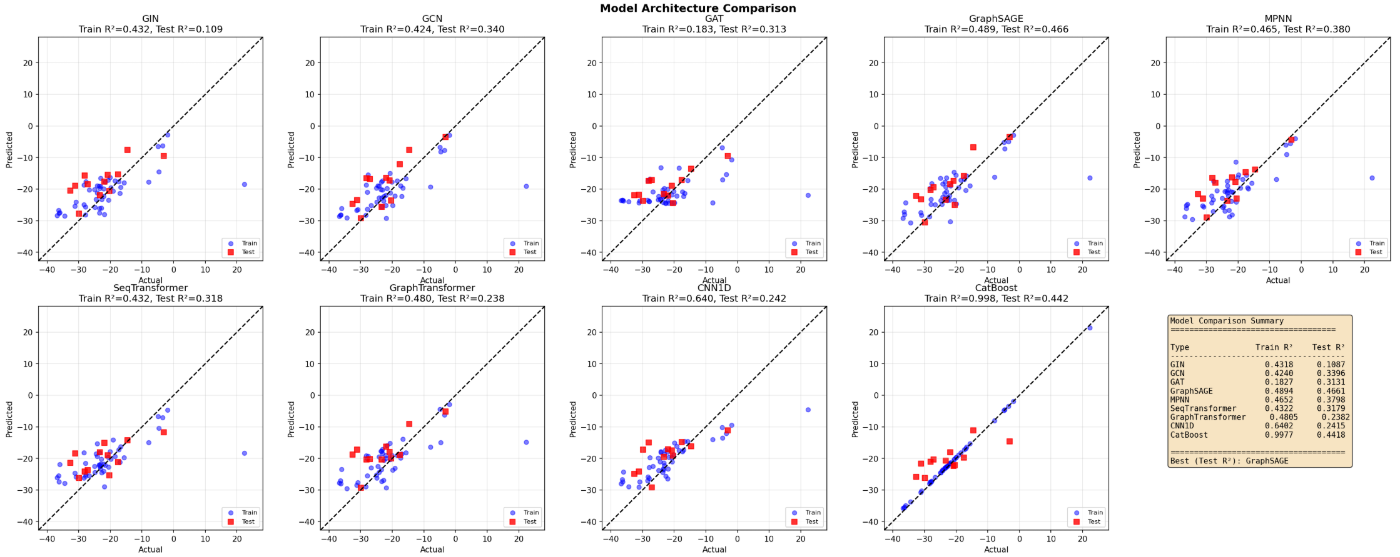
1. グラフニューラルネットワーク (GNN) 系化合物を「原子=ノード」「結合=エッジ」とするグラフ構造として捉えるモデル群です。
- GIN / GCN / GAT: グラフ上のノード情報を近傍から集約する標準的な手法。画像ではGATの精度が低く、このタスクへの適性は慎重な判断が必要です。
- GraphSAGE: 近傍ノードのサンプリングと集約を行う手法。
- MPNN (Message Passing Neural Network): 原子間のメッセージ伝達を模した、化学分野で非常に強力なモデルです。
2. トランスフォーマー (Transformer) 系自然言語処理で使われる「注目機構(Attention)」を応用したモデルです。
- SeqTransformer: ペプチドのアミノ酸配列(文字列情報)をシーケンスとして処理します。
- GraphTransformer: グラフ構造にAttention機構を導入し、原子同士の遠距離の相関も考慮します。
3. その他(CNN・勾配ブースティング)
- CNN1D: 1次元の畳み込みニューラルネットワーク。主に配列情報の局所的なパターン抽出に使用されます。
- CatBoost: 決定木ベースの勾配ブースティングアルゴリズム。Train $R^2 = 0.998$ と非常に高いですが、Testスコアとの乖離が大きく、過学習(Overfitting)の傾向が見て取れます。
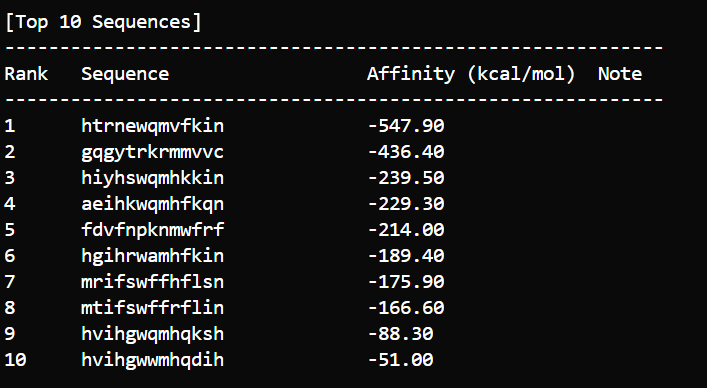
ペプチドの探索
モデルを選ぶと、
- さらに結合力が高い配列が提案(5個)
- 実際に分子ドッキング
- モデルの学習へ
- 1-4のループ
がなされる。最終的に結合力の高い配列が提案されます。
最終結果で結合力が高すぎるものはFalse positiveな感じもしますが、出てきたものについては、PRODIGYや実験でValidationするとよいでしょう。
最後に
以前紹介したRFPeptide→ProteinMPNNでDe novo環状ペプチドの合成した後の配列を使用すれば、
さらなる結合力の向上も可能です。ぜひ試してみてください!
今後は非天然アミノ酸の導入や膜透過性の指標も入れて、様々な指標でよい結果がなるような環状ペプチドの合成を試そうと思います!
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

