

本記事では、FEgrowというインシリコ創薬ツールとアクティブラーニング(Active Learning, AL)を組み合わせ、in silico screeningで得られた化合物を基にさらに効率よくスクリーニングしていく方法を紹介します。
【この記事のまとめ】
創薬研究者やバイオインフォマティクスエンジニアに向けて、インシリコ創薬ツール「FEgrow」と能動学習(アクティブラーニング)を組み合わせ、膨大な化学空間から有望な化合物を最小限の計算リソースで効率的に探索する手法を解説します。
- FEgrowによる分子設計と評価の自動化:足場分子(スキャフォールド)に対して多様なリンカーや置換基を結合させ、ドッキングシミュレーションを通じて結合親和性を評価するプロセスを習得できます。
- アクティブラーニングによるスクリーニングの高速化:全ての化合物候補を評価するのではなく、機械学習が「次に評価すべき化合物」を自律的に判断することで、計算時間を大幅に短縮しつつ高活性化合物へ到達する方法を紹介します。
- Google Colab/ローカル環境での実践ワークフロー:ライブラリのインストールから、SARS-CoV-2 Mproを標的とした具体的なチュートリアルコードの実行手順まで、即座に業務・研究へ応用できる形で網羅しています。
この記事を読むことで、計算コストの壁を突破し、限られた時間内でより高品質なリード化合物を特定するための高度なスクリーニング戦略を実装できるようになります。
動作検証済み環境
Windows 11 Home, 13th Gen Intel(R) Core(TM) i7-13700,
64 ビット オペレーティング システム、x64 ベース プロセッサ, メモリ:32GB

自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています



自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!


FEgrowとは?
FEgrowは、フラグメント分子(足場となる小さな分子)に、多様なリンカーや置換基(R-groups)を結合させることで、官能基を置換した化合物ライブラリを仮想的に構築し、その中から有望な化合物を効率的に設計・評価するためのツールです。
今回は、FEgrowのチュートリアルをフォローします。
新型コロナウイルスのメインプロテアーゼ(SARS-CoV-2 Mpro)を標的とした仮想的な化学空間(Chemical Space)から、予測される結合親和性(pK)を最適化する化合物を優先的に選定する過程を追っていきます。
アクティブラーニング(Active Learning)とは?
アクティブラーニング(AL)は、機械学習の一種で、「学習データが不足している状況下で、どのデータ(化合物)を優先的に評価すれば、最も効率よくモデルの精度を高められるか?」を自律的に判断する手法です。
創薬研究におけるALの役割は以下の通りです。
- 初期評価(ランダムサンプリング): まず少数の化合物をランダムに選び、その結合親和性を計算(評価)します。
- モデル構築: 評価結果を基に、未評価の化合物群のスコア(結合親和性)を予測する機械学習モデルを構築します。
- 情報量に基づく選択: モデルの予測結果から、「最もスコアが高そう」または「予測の不確実性が高く、評価すればモデルが大きく改善しそう」な化合物を、優先的に選択します。
- 再評価と学習: 選択した化合物を実際に評価し、その結果をモデルにフィードバック(学習)させることで、モデルの予測精度を向上させます。
このサイクルを繰り返すことで、全化合物を評価せずとも、最終的に目的とする**高性能な化合物(高い結合親和性を持つ化合物)に効率よくたどり着くことが可能になります。
FEgrowは、このALの仕組みを、化合物の設計・ドッキング評価プロセスに組み込むことを可能にしています。
環境構築
installationの資料はこちら。
以下をターミナルで打ちます。
# Miniforge3インストーラをダウンロード
# uname コマンドでOSとアーキテクチャ名を取得し、適切なファイル名を構築
curl -L -O "https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-$(uname)-$(uname -m).sh"
# Miniforge3をインストール
bash Miniforge3-$(uname)-$(uname -m).sh
# 👇ここからconda環境を初期化・設定
# Condaの初期設定を行うためのフックを現在のシェルセッションにロード
# /home/shizuku/miniforge3/ はMiniforge3のインストール先ディレクトリに合わせる
eval "$(/home/shizuku/miniforge3/bin/conda shell.bash hook)"
# bashシェルでcondaコマンドを適切に動作させるために初期化設定をシェル設定ファイル(例: ~/.bashrc)に書き込む
conda init bash
# シェル設定ファイル(.bashrc)を再読み込みし、上記の設定を現在のセッションに反映
source ~/.bashrc
# 👇ここからFEgrowのセットアップ
# FEgrowリポジトリをクローン(作業ディレクトリはどこでも可)
git clone https://github.com/cole-group/FEgrow.git
# クローンしたディレクトリに移動
cd FEgrow
# mamba(condaの高速版)を使って、environment.ymlファイルに基づいて仮想環境を作成
mamba env create -f environment.yml
# 作成した仮想環境 'fegrow' をアクティベート
conda activate fegrow
# FEgrowパッケージを依存関係なしでインストール(開発モードや、環境ファイルが依存関係を処理済みのため)
# 最後のピリオド '.' はカレントディレクトリ(FEgrowのルート)にあるセットアップファイルを参照
pip install --no-deps .
| コマンド | 説明 |
|---|---|
curl -L -O ... |
Miniforge3インストーラのダウンロード。 |
curl: URLからデータを転送するためのコマンドラインツール。-L: リダイレクトを辿る(GitHubの「latest」リンクに対応するため)。-O: ダウンロードしたファイルを、リモートファイル名と同じ名前で現在のディレクトリに保存する。$(uname)と$(uname -m): OS名(例:Linux,Darwin)とアーキテクチャ名(例:x86_64,arm64)をコマンド実行時に動的に取得し、適切なインストーラファイル名を構築する。 | |bash Miniforge3-$(uname)-$(uname -m).sh| Miniforge3のインストール。ダウンロードしたシェルスクリプトを実行し、Miniforge3をインストールする。 | |eval "$(/home/shizuku/miniforge3/bin/conda shell.bash hook)"| Condaの初期設定を現在のシェルセッションにロード。condaコマンドを一時的に利用可能にするための設定(フック)を現在のセッションに適用する。miniforge3のインストールパスは環境によって異なる可能性があります。 | |conda init bash| Condaの永続的な初期設定。condaコマンドを次回以降のシェル起動時にも使えるように、使用しているシェル設定ファイル(~/.bashrcなど)に設定を書き込む。 | |source ~/.bashrc| 設定ファイルの再読み込み。conda init bashで書き込まれた設定を現在のシェルセッションに反映させるために、設定ファイル(ここでは.bashrc)を再読み込みする。 | |git clone https://github.com/cole-group/FEgrow.git| リポジトリのクローン。指定されたURLからFEgrowのGitリポジトリを現在のディレクトリにダウンロードする。 | |cd FEgrow| ディレクトリの移動。クローンして作成されたFEgrowディレクトリに移動する。 | |mamba env create -f environment.yml| 仮想環境の作成。mamba(condaの高速な実装)を使用して、environment.ymlファイルに記述されている依存関係(パッケージ)を持つ仮想環境を作成する。 | |conda activate fegrow| 仮想環境の有効化。新しく作成されたfegrowという名前の仮想環境を現在のシェルセッションでアクティブにする。これにより、その環境内のPythonやパッケージが使えるようになる。 | |pip install --no-deps .| パッケージのインストール。カレントディレクトリ(.)にあるPythonパッケージ(FEgrow自体)をインストールする。pip: Pythonのパッケージ管理システム。--no-deps: 依存関係のチェックとインストールをスキップする(既にmamba env createで依存関係が処理されているため)。.: カレントディレクトリを参照し、setup.pyまたはpyproject.tomlに基づいてパッケージをインストールする。 |
クローンしてきたFEgrowのtutorialsの2_tutorial_active_learning.ipynb をcursorで開き、一つずつrunしてください。
cursorでのjupyter環境の設定は以下をご覧ください。
📰 環境構築|【完全版】In silico創薬実践書 〜おうち創薬で論文を書こう〜
FEgrowによるアクティブラーニングの実行
全コードはこちらを参考にしています。一部うまくいかなかった部分があるので、少し変更しています。チュートリアルとほぼ一緒ですが、GPUは使用しておりません。gnina_gpu =False にして実行しています。
# ----------------------------------------------------
# STEP 1: 準備と環境設定
# ----------------------------------------------------
# 必要なライブラリをインポート
import prody # タンパク質(受容体)の操作・解析用ライブラリ
from rdkit import Chem # 分子構造(化学)の操作用ライブラリ
import fegrow # 分子の拡張とドッキング(結合予測)を行う中心ライブラリ
# fegrow内の主要なクラスをインポート
from fegrow import ChemSpace, Linkers, RGroups
from fegrow.al import Model, Query # Active Learning(能動学習)のモデルとクエリをインポート
rgroups = RGroups() # 利用可能なRグループ(置換基)のコレクションを準備
linkers = Linkers() # 利用可能なリンカー(つなぎ)のコレクションを準備
# DaskのLocalClusterを設定し、計算を並列化(複数のCPUコアで同時に実行)できるようにする
from dask.distributed import LocalCluster
lc = LocalCluster(processes=True, n_workers=None, threads_per_worker=1)
# create the chemical space (化学空間、つまり作成する可能性のある分子の集合) を作成
cs = ChemSpace(dask_cluster=lc) # Daskクラスターと関連付ける
cs
# turn on the caching in RAM (optional)
# 計算結果をメモリ(RAM)にキャッシュし、処理を速くする(任意)
cs.set_dask_caching()
# ----------------------------------------------------
# STEP 2: 足場分子とターゲットタンパク質の準備
# ----------------------------------------------------
# 足場(コアとなる分子骨格)の構造ファイル(.sdf)を読み込む
init_mol = Chem.SDMolSupplier("sarscov2/5R83_core.sdf", removeHs=False)[0]
# rdkitのMolオブジェクトをfegrow特有のRMol形式に変換
scaffold = fegrow.RMol(init_mol)
# 足場の2D画像を表示(idx=Trueで原子番号を表示、サイズを指定)
scaffold.rep2D(idx=True, size=(500, 500))
# specify the attachment point (in this case hydrogen atom number 6)
# どこに新しい部分をつなぐか(ここでは原子番号6の水素の位置)を指定
attachmentid = 6
# 接続ポイント(原子番号6)の原子をダミー原子(原子番号0、通常は'*')に置き換える
scaffold.GetAtomWithIdx(attachmentid).SetAtomicNum(0)
cs.add_scaffold(scaffold) # 処理した足場を化学空間に追加
# load the receptor structure
# ターゲットタンパク質(受容体)の構造ファイル(.pdb)を読み込む
sys = prody.parsePDB("sarscov2/5R83_final.pdb")
# remove any unwanted molecules
# 受容体から核酸、ヘテロ原子(cofactorなど)、水分子を除外して純粋なタンパク質を抽出
rec = sys.select("not (nucleic or hetatm or water)")
# save the processed protein
# 処理済みのタンパク質を新しいファイルに保存
prody.writePDB("rec.pdb", rec)
# fix the receptor file (missing residues, protonation, etc)
# 受容体ファイルに残基の欠損修正やプロトン化状態の調整などを行う
fegrow.fix_receptor("rec.pdb", "rec_final.pdb")
cs.add_protein("rec_final.pdb") # 最終的な受容体(タンパク質)を化学空間に追加
# ----------------------------------------------------
# STEP 3: 化学空間の構築(分子候補の網羅的な作成)
# ----------------------------------------------------
numlinkers = 50 # 試すリンカーの数
numrgroups = 50 # 試すRグループの数
# 指定された数のリンカーとRグループの全ての組み合わせを作成し、化学空間に追加する
for i in range(numlinkers):
if i % 10 == 0:
print(i) # 進行状況を10個ごとに表示
for j in range(numrgroups):
# リンカー[i]とRグループ[j]を組み合わせて化学空間に分子の候補を追加
cs.add_rgroups(linkers.Mol[i], rgroups.Mol[j])
# The chemical space now includes our 2500 small molecules:
# 化学空間に分子候補の総数(50x50=2500)が追加された
cs
# 化学空間の最初の分子(cs[0])の2D構造を表示
cs[0].rep2D()
# ----------------------------------------------------
# STEP 4: 能動学習(Active Learning)の初期評価
# ----------------------------------------------------
# Pick 50 random molecules
# Active Learning(能動学習)の最初のステップとして、ランダムに50個の分子を選ぶ
random1 = cs.active_learning(50, first_random=True)
# now evaluate the first selection, note that dask is used to parallelise the calculation
# 選ばれた50個の分子に対してドッキングシミュレーションを実行
# daskが並列計算を行う
# molecules that cannot be built assigned a predicted affinity of 0
# gnina_gpu=Falseに変更 (GPUを使わずにCPUでgninaを実行することを指定)
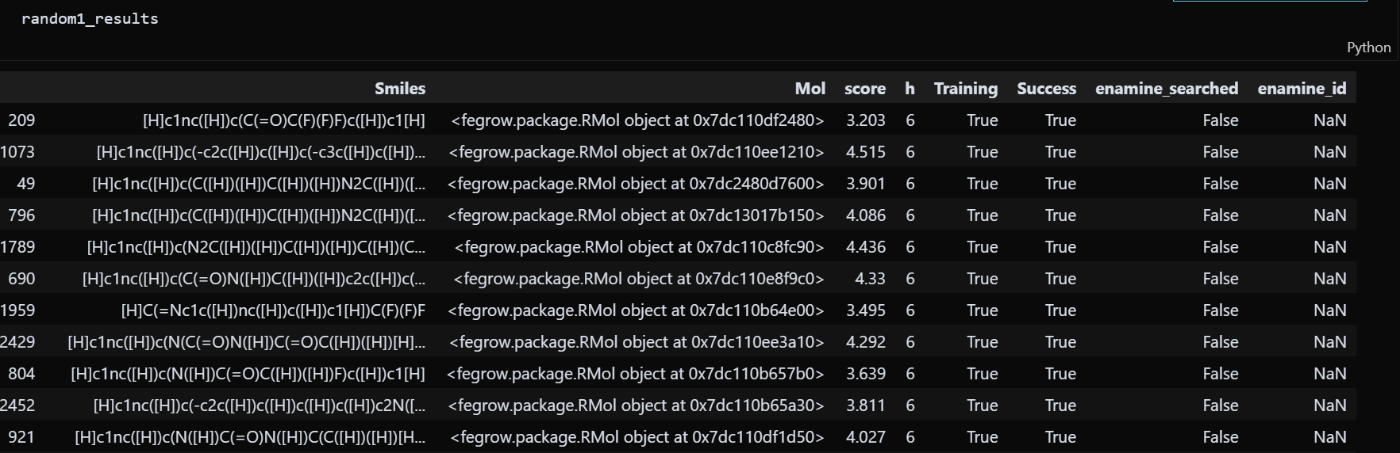
random1_results = cs.evaluate(
random1, num_conf=50, gnina_gpu=False, penalty=0.0, al_ignore_penalty=False
)
random1_results # 結果(ドッキングスコアなど)を表示
# スコア(計算値)がある(NaNではない)分子だけを抽出
computed = cs.df[~cs.df.score.isna()]
print("Computed cases in total: ", len(computed)) # 計算された分子の総数を表示
# ----------------------------------------------------
# STEP 5: AIモデルの学習と最適分子の選択
# ----------------------------------------------------
# チュートリアルにはない部分。付け足しました。
# gninaのパスを明示的に設定
import os
from pathlib import Path
# 現在のディレクトリのgninaバイナリを設定(セキュリティや環境依存性のため)
gnina_path = os.path.abspath("gnina")
if os.path.exists(gnina_path):
fegrow.RMol.set_gnina(gnina_path) # gnina実行ファイルの場所をfegrowに教える
print(f"gnina path set to: {gnina_path}")
else:
# gninaが見つからない場合はメッセージを表示
print(f"gnina not found at: {gnina_path}")
print("Available files in current directory:")
for f in os.listdir("."):
if "gnina" in f.lower():
print(f" - {f}")
# AIの学習モデルとしてガウス過程を設定
cs.model = Model.gaussian_process()
# 分子選択の戦略としてUCB (Upper Confidence Bound) を設定(探索と利用のバランスをとる)
cs.query = Query.UCB(beta=1)
# gnina_gpu=Falseでないとうまくいかない。 # 環境によってはGPUの使用がうまくいかないため、CPUに固定する
for cycle in range(3): # 能動学習のプロセスを3回繰り返す
print(f"Starting cycle {cycle + 1}/3...") # 現在のサイクル数を表示
try:
# AIモデルと選択戦略に基づいて、次に計算すべき最適な50個の分子を選ぶ
picks = cs.active_learning(50)
print(f"Selected {len(picks)} molecules for evaluation") # 選ばれた分子数を表示
# 選ばれた50個の分子をドッキングシミュレーションで評価
picks_results = cs.evaluate(
picks, num_conf=50, gnina_gpu=False, penalty=0.0, al_ignore_penalty=False
# num_conf=50: 分子の立体構造(コンフォメーション)を50個生成して試す
# gnina_gpu=False: GNINAをCPUで実行するように指定
)
# save the new results
# 今回の結果をサイクルごとに異なるCSVファイルに保存
picks_results.to_csv(f"notebook_iteration{cycle}_results.csv")
print(f"Cycle {cycle + 1} completed successfully. Results saved to notebook_iteration{cycle}_results.csv")
except Exception as e:
# エラーが発生した場合、メッセージを表示し、次のサイクルへ進む
print(f"Error in cycle {cycle + 1}: {str(e)}")
print("Continuing with next cycle...")
continue
cs.model = Model.gaussian_process() # Active Learning(能動学習)のモデルとしてガウス過程を設定
cs.query = Query.Greedy() # 次に評価する分子の選択戦略としてGreedy(貪欲法)を設定
# モデルを使って予測し、次に計算すべき最適な50個の分子を選ぶ
picks = cs.active_learning(50)
# 選ばれた50個の分子をドッキングシミュレーションで評価
picks_results = cs.evaluate(
picks, num_conf=50, gnina_gpu=False, penalty=0.0, al_ignore_penalty=False
)
# save the new results
picks_results.to_csv("notebook_greedy_results.csv") # 結果をCSVファイルに保存
# ----------------------------------------------------
# STEP 6: 結果の統合と上位分子の抽出・保存
# ----------------------------------------------------
# save the chemical space of built molecules:
# 構築された(または構築できなかった)全ての分子を含む化学空間を保存する準備
failed = False
unbuilt = False
# 結果をSDファイル(分子構造とそのプロパティを格納するファイル形式)として書き出す
with Chem.SDWriter("notebook_chemspace.sdf") as SD:
# 分子構造以外のプロパティ(スコア、成功フラグなど)の列名リストを作成
columns = cs.df.columns.to_list()
columns.remove("Mol")
# 化学空間内の全ての行(分子候補)をループ処理
for i, row in cs.df.iterrows():
# ignore this molecule because it failed during the build
# 構築に失敗した分子をスキップする設定(failed=Falseなのでスキップしない)
if failed is False and row.Success is False:
continue
# ignore this molecule because it was not built yet
# 未構築の分子をスキップする設定(unbuilt=Falseなので、SuccessがFalseならスキップしない)
if unbuilt is False and not row.Success:
continue
mol = row.Mol # RDKit Molオブジェクトを取得
mol.SetIntProp("index", i) # 元のインデックスをプロパティとして設定
for column in columns: # 各プロパティをMolオブジェクトに設定
value = getattr(row, column)
mol.SetProp(column, str(value))
mol.ClearProp("attachement_point") # 不要なプロパティをクリア
SD.write(mol) # SDファイルに分子構造とプロパティを書き込む
# save the structures of the top 10 molecules in ranked order as a sdf file:
# ドッキングスコアの高い上位10個の分子を保存する処理
molecules = []
input_sdf = "notebook_chemspace.sdf" # 保存したSDファイル
best_n = 10 # 上位10個
with Chem.SDMolSupplier(input_sdf) as SDF:
# for each mol
# 保存したSDファイルから分子を一つずつ読み込む
for mol in SDF:
if mol is None: # 読み込めなかったらスキップ
continue
# 構築に成功した分子('Success'プロパティが'True')のみをリストに追加
if mol.GetPropsAsDict()["Success"] == "True":
molecules.append(mol)
# sort by the key
# 分子リストを、プロパティの'score'の値(ドッキングスコア)で降順(reverse=True、高いスコアが先)にソート
sorted_molecules = sorted(
molecules, key=lambda m: m.GetPropsAsDict()["score"], reverse=True
)
# ソートされた分子の上位10個を新しいSDファイルに書き出す
with Chem.SDWriter(f"top_{best_n:d}_{input_sdf}") as SDF_OUT:
for i, mol in enumerate(sorted_molecules):
if i == best_n: # 10個書き出したら終了
break
SDF_OUT.write(mol)
print("Done") # 処理完了を通知
全体の流れ:AIを使った新しい薬の候補探し
このコードは、既存の薬の骨格をベースに、AI(機械学習)を使って、ターゲットタンパク質に最もよく結合するであろう新しい分子(薬の候補)を効率的に見つけ出すという、創薬研究のプロセスをシミュレーションしています。
プロセスは以下のステップで進行します。
- 準備と分子設計(Step 1〜3):
- 必要なツールを準備し、PCの並列処理環境を立ち上げます。
- 薬のコア骨格とターゲットタンパク質を読み込み、シミュレーション用に加工します。
- コア骨格に50種のリンカーと50種のRグループを組み合わせ、2,500個の分子候補(化学空間)を作成します。
- 初期評価(Step 4):
- 作成した分子候補からランダムに50個を選び出し、ドッキングシミュレーション(結合予測)を実行してスコアを計算します。これはAIが学習するための最初のデータセットとなります。
- AI学習と最適化(Step 5):
- 初期評価の結果を元に、AIモデル(ガウス過程)が学習します。
- AIは学習結果に基づき、「次に計算すれば最も良いスコアが出そう」と予測した最適な50個の分子を選び出し、再度シミュレーションを実行します。
- 結果の統合と抽出(Step 6):
- 全ての分子の構造と評価スコアを一つのファイルに統合して保存します。
- 最終的に、全分子の中からドッキングスコアが最も高い上位10個の分子を抜き出し、最終ファイルとして保存します。
STEP 1: 準備と環境設定
シミュレーションを行うための道具の準備と、計算を効率化するための設定を行います。
import prody/from rdkit import Chem/import fegrow: タンパク質操作のProDy、分子構造操作のRDKit、そして創薬シミュレーションの中心となるfegrowという専門的なツールキットをプログラムに組み込んでいます。from fegrow import ChemSpace, Linkers, RGroups:fegrowから、分子リストの管理者(ChemSpace)、部品カタログのデータ(Linkers,RGroups)といった、主要な機能を呼び出せるようにしています。rgroups = RGroups()/linkers = Linkers(): Rグループとリンカーの**利用可能なリスト(部品カタログ)**を、プログラム内で実際に使える形に初期化しています。lc = LocalCluster(...): Daskというライブラリを使い、あなたのPCの複数のCPUコアを計算に使えるようにするための**並列処理チーム(LocalCluster)**を立ち上げています。これにより、計算が速くなります。cs = ChemSpace(dask_cluster=lc): これから作る全ての分子の構造やスコアを管理するメインのワークシート(化学空間)を作成し、並列処理チーム(lc)と連携させています。cs.set_dask_caching(): 計算結果をメモリに一時保存し、同じ計算を省略することで処理を高速化する設定を有効にしています。
STEP 2: 足場分子とターゲットタンパク質の準備
新しい薬のコアとなる骨格と、結合させるターゲット(受容体)をシミュレーションに適した形に加工し、登録します。
-
init_mol = Chem.SDMolSupplier("...")[0]: 薬のコア骨格(足場)の構造ファイル(.sdf)を読み込みます。 -
scaffold = fegrow.RMol(init_mol): 読み込んだ分子データを、fegrow専用の構造形式(RMol)に変換しています。 -
scaffold.rep2D(idx=True, size=(500, 500)): 足場分子の2D構造図を、原子番号付きで表示します。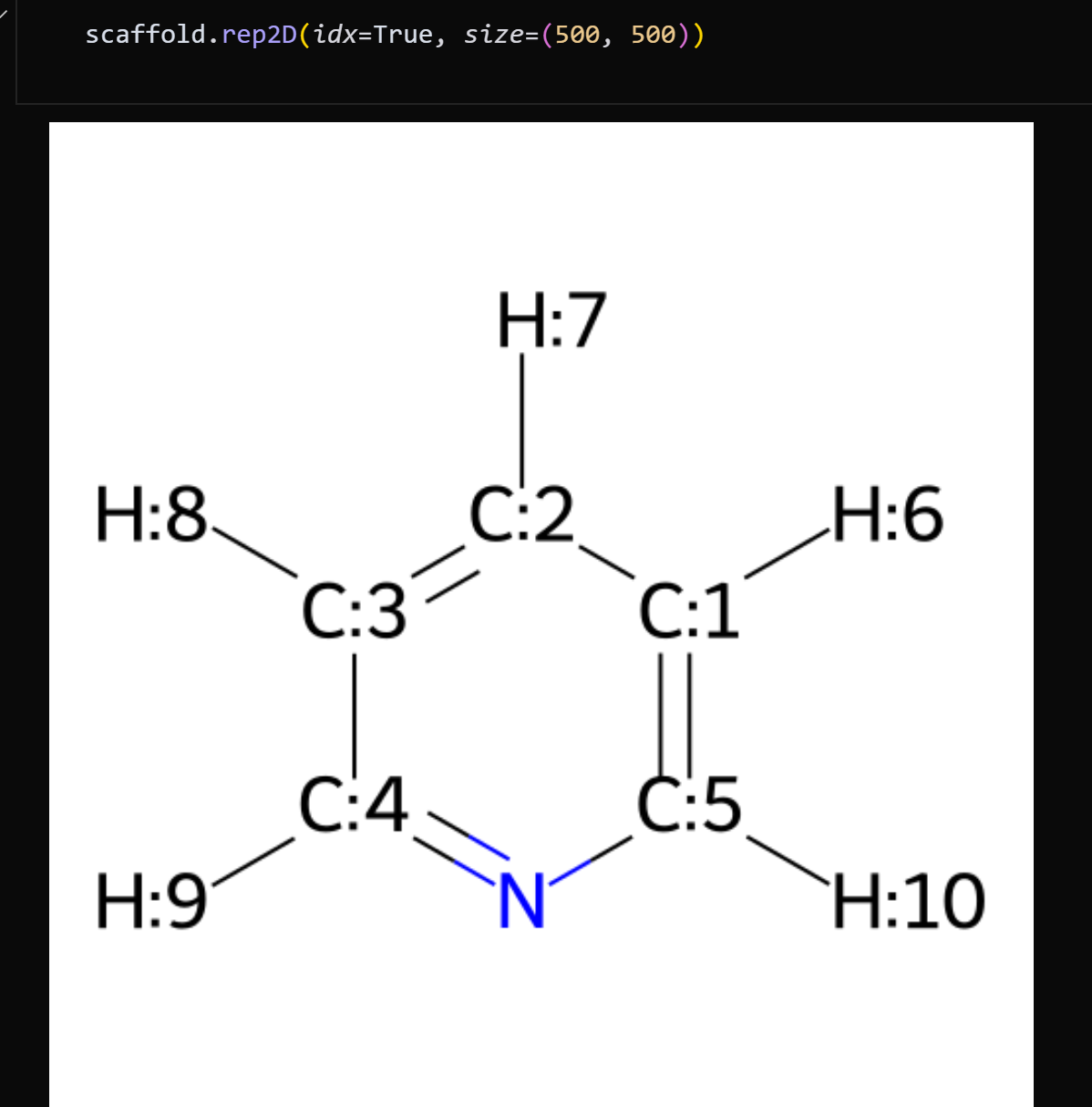
-
attachmentid = 6: 新しい部品をつなげる場所として、原子番号6の原子を指定しています。 -
scaffold.GetAtomWithIdx(attachmentid).SetAtomicNum(0): 指定した結合点(原子番号6)を、ダミー原子('*')という**「ここに部品をつなぐ目印」**に置き換えています。 -
cs.add_scaffold(scaffold): 加工済みの足場分子を化学空間(cs)に登録します。 -
sys = prody.parsePDB("..."): 薬が結合するターゲットタンパク質の構造データ(.pdbファイル)を読み込みます。 -
rec = sys.select("not (nucleic or hetatm or water)"): タンパク質構造から、核酸、水、不純物といったシミュレーションに不要なものを取り除いています。 -
fegrow.fix_receptor("rec.pdb", "rec_final.pdb"): 一時ファイル(rec.pdb)に対し、欠損修正やプロトン化を行い、最終的な受容体ファイル(rec_final.pdb)を作成しています。 -
cs.add_protein("rec_final.pdb"): 最終調整を終えたターゲットタンパク質を化学空間(cs)に登録し、シミュレーションの準備を完了します。 -
attachmentid = 6: 新しい部品をつなげる場所として、原子番号6の原子を指定しています。 -
scaffold.GetAtomWithIdx(attachmentid).SetAtomicNum(0): 指定した原子(原子番号6)を、「ここに部品をつなぐ」という**目印(ダミー原子)**に置き換えています。H:6に様々な官能基を伸ばしていくという設定を行っています。 -
cs.add_scaffold(scaffold): 処理を終えた足場分子を、化学空間(ワークシートcs)に登録します。 -
sys = prody.parsePDB("..."): ターゲットタンパク質(受容体)の構造データ(.pdbファイル)を読み込みます。 -
rec = sys.select("not (nucleic or hetatm or water)"): タンパク質の構造から、核酸、水分子、不純物(hetatm)などのシミュレーションに不要なものを取り除き、純粋なタンパク質を選び出しています。 -
fegrow.fix_receptor("rec.pdb", "rec_final.pdb"): 選出したタンパク質構造に対し、欠損部分の補完やプロトン化状態の調整などを行い、シミュレーションに適した最終ファイル(rec_final.pdb)を作成しています。 -
cs.add_protein("rec_final.pdb"): 最終的に準備されたターゲットタンパク質を、化学空間(cs)に登録します。
STEP 3: 化学空間の構築(分子候補の網羅的な作成)
足場に全ての部品の組み合わせを適用し、総勢2,500個の分子候補を作成します。
-
numlinkers = 50/numrgroups = 50: 使用するリンカーとRグループの数をそれぞれ50個に設定しています。 -
for i in range(...)/for j in range(...): 二重ループを使い、50個のリンカーと50個のRグループの**全ての組み合わせ(50×50=2,500通り)**を順に処理する指示です。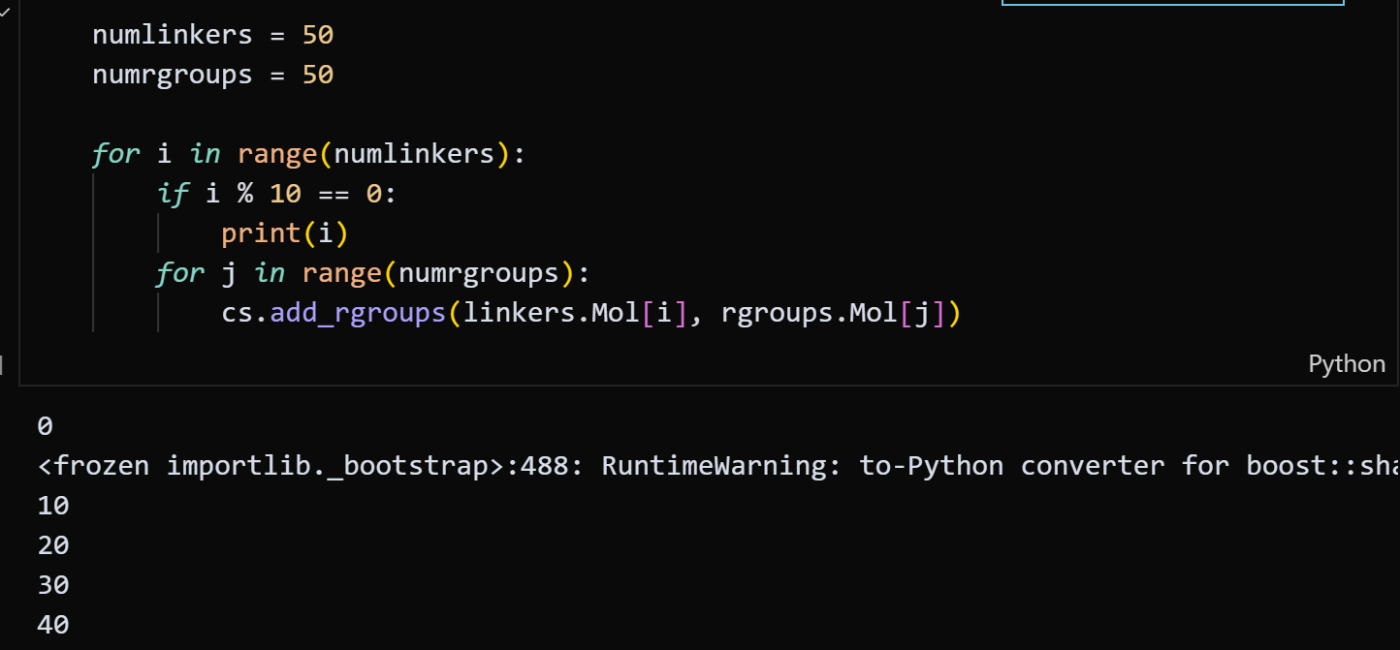
-
cs.add_rgroups(linkers.Mol[i], rgroups.Mol[j]): ループで選ばれた特定のリンカーとRグループを足場につなげた新しい分子候補を、化学空間(cs)に登録しています。
-
cs[0].rep2D(): 作成された分子候補リストの最初の分子の2D構造を表示し、正しく分子が構築されているかを確認します。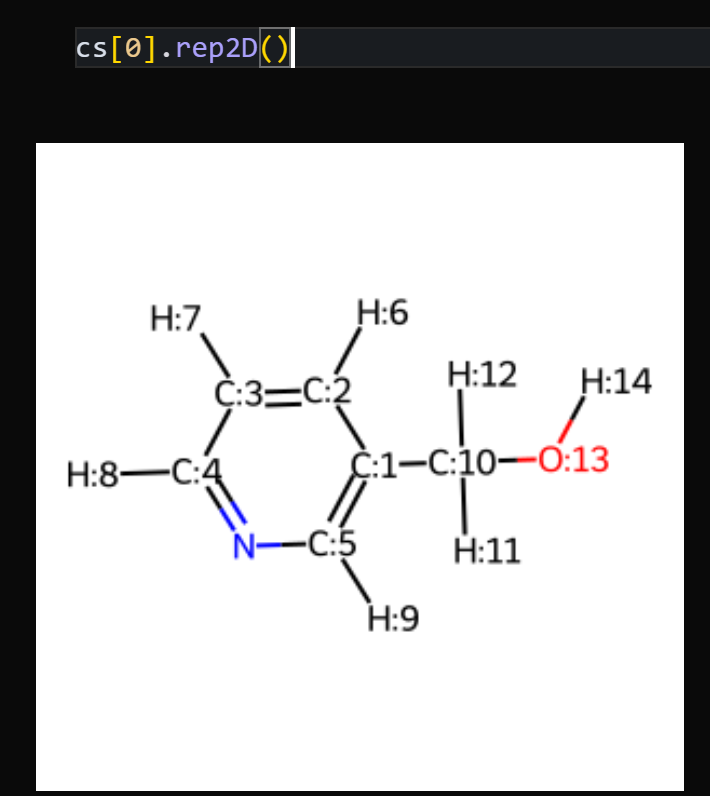
STEP 4: 能動学習(Active Learning)の初期評価
AIに学習させるための初期データを収集するため、ランダムに分子を選んでシミュレーションを実行します。
random1 = cs.active_learning(50, first_random=True): 能動学習の初期段階として、ランダムに50個の分子を選び出します。random1_results = cs.evaluate(random1, ...): 選ばれた50個の分子について、ドッキングシミュレーション(結合予測)を実行し、スコアを計算しています。gnina_gpu=FalseはCPUでの計算を意味します。
以下のような出力になります。長い出力です。公式のマークダウンを合わせて参考にしてください。
random1_results: 計算が完了した初期評価の結果(スコアなどが含まれる表形式のデータ)を表示しています。
computed = cs.df[~cs.df.score.isna()]: スコアが計算できた(NaNではない)分子だけを抽出し、計算が成功した数を数えています。
STEP 5: AIモデルの学習と最適分子の選択
初期データでAIが学習し、次に試すべき最適な分子を選び出して最終評価を行います。
fegrow.RMol.set_gnina(gnina_path): ドッキングシミュレーションに使うGNINAというプログラムがどこにあるか(パス)をfegrowに明示的に設定しています。cs.model = Model.gaussian_process(): 能動学習のためのAIの学習モデルとして、ガウス過程という予測手法を設定しています。cs.query = Query.UCB(beta=1)Queryは、次にどの分子を評価するかを決める戦略(クエリ戦略)を設定しています。- UCB(Upper Confidence Bound:上限信頼限界)は、「探索(Exploration)」と「利用(Exploitation)」という2つの行動をバランス良く行うための戦略です。
- 利用: AIが「この分子はスコアが高そう(良い結果が得られそう)」と予測する分子を選ぶことです。
- 探索: AIが「この分子のことはよくわからない(不確実性が高い)」と判断する分子を、予測スコアに関わらず選ぶことです。
- UCB戦略は、「予測スコア+不確実性の幅」が大きい分子を選びます。これにより、単に予測スコアが高い分子だけでなく、まだ情報が少なく「もしかしたらすごく良い分子かもしれない」未知の領域にも積極的に挑戦できます。
cs.query = Query.Greedy(): AIが次に試す分子を選ぶ際の戦略として、Greedy(貪欲法)(最も期待値の高いものを選択する戦略)を設定しています。picks = cs.active_learning(50): AIが予測した結果に基づき、「最も高いスコアが出そう」な最適な50個の分子を選び出します。picks_results = cs.evaluate(picks, ...): AIが選んだ50個の分子に対し、再度ドッキングシミュレーションを実行してスコアを計算します。picks_results.to_csv("notebook_greedy_results.csv"): 2回目の評価結果を、CSVファイルとして保存しています。
STEP 6: 結果の統合と上位分子の抽出・保存
全ての分子のデータを整理し、最も優秀な分子を抽出して最終ファイルとして保存します。
with Chem.SDWriter("notebook_chemspace.sdf") as SD:: 全ての分子の構造と評価結果を記録するためのSDファイルの書き出しを開始します。for i, row in cs.df.iterrows():: ワークシート(cs.df)にある全分子を一つずつ処理するためのループです。mol.SetProp(column, str(value)): 分子の構造データに対し、計算で得られたスコアや成功/失敗フラグなどの情報を**プロパティ(付箋)**として書き込んでいます。SD.write(mol): 情報が付加された分子構造をSDファイルに書き込み、全分子のデータ統合を完了させています。best_n = 10: 最終的に抽出する上位分子の数を10個に設定しています。if mol.GetPropsAsDict()["Success"] == "True":: 構築に成功した分子だけを選び出すための条件です。sorted_molecules = sorted(molecules, key=lambda m: ..., reverse=True): 成功した分子のリストを、ドッキングスコア(score)が高い順に並び替えています。with Chem.SDWriter(f"top_{best_n:d}_...") as SDF_OUT:: 並び替えたリストの上位10個の分子だけを、新しいSDファイルとして書き出し、最終的な成果物として保存しています。print("Done"): すべての処理が完了したことを知らせるメッセージです。
結果の可視化
もともとのタンパク質と出力されたtop_10_notebook_chemspace.sdf をpymolにドラック&ドロップして表示されると上位10個の結合力の高い化合物が表示されます。右下の再生ボタンを押せば各分子の結合の様式がわかります。
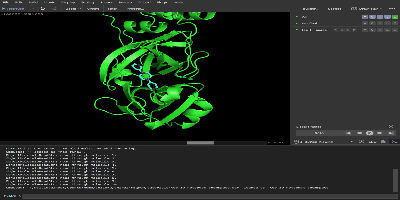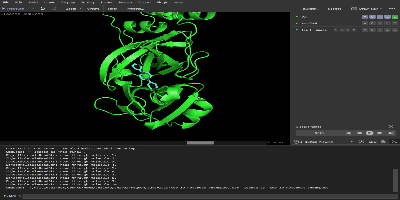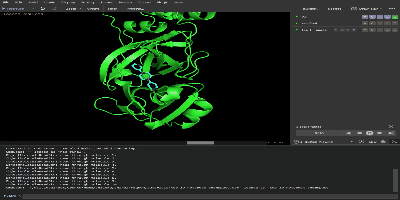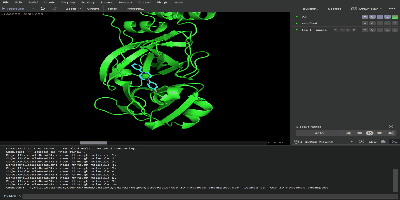
最後に
FEgrowとアクティブラーニング(AL)を活用したシミュレーション、いかがでしたでしょうか。
数千にも及ぶ大規模な仮想ライブラリの中から、わずか数回のサイクルで最も結合親和性が高いと予測される化合物を効率的に見つけ出すことができました。
全ての仮想ライブラリを網羅的に計算することは、膨大な計算リソースと時間が必要となり現実的ではありません。しかし、ALは初期の限られたデータからAIが学習し、次に試すべき最適な分子を予測することで、リソースを大幅に節約しながら、有望な化合物に素早くたどり着くための強力な探索手法となります。
ぜひ皆さんもFEgrowとアクティブラーニングを組み合わせ、効率的なインシリコ創薬に取り組んでみてください!
参考文献
GitHub – cole-group/FEgrow: An Open-Source Molecular Builder and Free Energy Preparation Workflow
License: MIT, Apache-2.0
自宅でできるin silico創薬の技術書を販売中
新薬探索を試したい方必読!
ITエンジニアである著者の視点から、wetな研究者からもdryの創薬研究をわかりやすく身近に感じられるように解説しています
自宅でできるin silico創薬の技術書を販売中
タンパク質デザイン・モデリングに焦点を当て、初めてこの分野に参入する方向けに、それぞれの手法の説明から、環境構築、実際の使い方まで網羅!

