

今回は、RNA-seq解析の精度を高めるために欠かせない「遺伝子アノテーション情報の活用方法」について詳しく解説します。すでに前処理されたアノテーションデータを用いて、遺伝子の基本情報を確認する方法から、転写因子の抽出分析、発現データとの統合解析までをステップごとに紹介します。この記事を通して、遺伝子アノテーションをうまく活用することで、RNA-seq解析の理解と発見がぐっと深まることを実感していただけるはずです。
macOS Monterey (12.4), Quad-Core Intel Core i7, Memory 32GB


公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!

公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!


TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます
解析準備
GEOからカウントデータを取得
今回の解析に使うカウントデータを取得してください。
# GEOqueryパッケージを読み込む
library(GEOquery)
# GEOからカウントデータを取得
gse <- getGEO("GSE141413", GSEMatrix = FALSE)
# カウントデータを含む補足ファイルをダウンロード
supp_dir <- "data/geo_suppl"
dir.create(supp_dir, recursive = TRUE, showWarnings = FALSE)
count_file <- getGEOSuppFiles("GSE141413", baseDir = supp_dir, makeDirectory = FALSE)
# ダウンロードしたカウントデータを読み込む
counts_path <- file.path(supp_dir, "GSE141413_gene_count.txt.gz")
counts_data <- read.delim(counts_path, header = TRUE)カウントデータの前処理
取得したカウントデータを解析に適した形式に整理します。このデータセットには発現量データと遺伝子アノテーション情報が含まれています。
# ステップ1: カウントデータの確認
dim(counts_data)
head(counts_data[, 1:8])
# ステップ2: データの構造確認
colnames(counts_data)
str(counts_data)
# ステップ3: 発現データとアノテーション情報の分離
count_cols <- c("Control", "NC_LM", "st2_LM", "IFNG", "st2_IFNG")
annotation_cols <- c("gene_name", "gene_chr", "gene_start", "gene_end", "gene_strand", "gene_length", "gene_biotype", "gene_description", "tf_family")
# サブセットの作成
counts <- counts_data[, count_cols]
gene_annotation <- counts_data[, annotation_cols]
# ステップ4: 行名の設定
rownames(counts) <- rownames(counts_data)
rownames(gene_annotation) <- rownames(counts_data)
# ステップ5: サンプル情報の作成
sample_info <- data.frame( sample_id = colnames(counts), condition = c("Control", "NC_LM", "st2_LM", "IFNG", "st2_IFNG"), group = c("Control", rep("LM", 2), rep("IFNg", 2)), treatment = c("None", "NC", "st2", "None", "st2"), row.names = colnames(counts)
)
# ステップ6: フィルタリングとデータ保存
# 空でない遺伝子のみを保持
counts <- counts[rowSums(counts) > 0, ]
gene_annotation <- gene_annotation[rownames(counts), ]これで準備完了です。
遺伝子アノテーション情報の活用方法
RNA-seq解析では、遺伝子の機能情報を活用することが重要です。ここでは、すでに処理した遺伝子アノテーション情報を使って様々な分析を行います。
遺伝子アノテーションの確認
# 前処理済み遺伝子アノテーションの読み込み
gene_annotation <- read.csv("data/processed/gene_annotation.csv", row.names = 1)# アノテーション情報の確認
head(gene_annotation)
summary(gene_annotation)出力例は以下になります。
> head(gene_annotation) gene_name gene_chr gene_start gene_end gene_strand gene_length
ENSMUSG00000069516 Lyz2 10 117277331 117282321 - 1316
ENSMUSG00000024661 Fth1 19 9982703 9985092 + 866
ENSMUSG00000029816 Gpnmb 6 49036546 49070929 + 6514
ENSMUSG00000007891 Ctsd 7 142375911 142388038 - 2351
ENSMUSG00000021939 Ctsb 14 63122462 63145923 + 6051
ENSMUSG00000004207 Psap 10 60277627 60302597 + 2744 gene_biotype
ENSMUSG00000069516 protein_coding
ENSMUSG00000024661 protein_coding
ENSMUSG00000029816 protein_coding
ENSMUSG00000007891 protein_coding
ENSMUSG00000021939 protein_coding
ENSMUSG00000004207 protein_coding gene_description
ENSMUSG00000069516 lysozyme 2 [Source:MGI Symbol;Acc:MGI:96897]
ENSMUSG00000024661 ferritin heavy polypeptide 1 [Source:MGI Symbol;Acc:MGI:95588]
ENSMUSG00000029816 glycoprotein (transmembrane) nmb [Source:MGI Symbol;Acc:MGI:1934765]
ENSMUSG00000007891 cathepsin D [Source:MGI Symbol;Acc:MGI:88562]
ENSMUSG00000021939 cathepsin B [Source:MGI Symbol;Acc:MGI:88561]
ENSMUSG00000004207 prosaposin [Source:MGI Symbol;Acc:MGI:97783] tf_family
ENSMUSG00000069516 -
ENSMUSG00000024661 -
ENSMUSG00000029816 -
ENSMUSG00000007891 -
ENSMUSG00000021939 -
ENSMUSG00000004207 -head(gene_annotation) の出力解説:
上記の出力は遺伝子アノテーションデータフレームの最初の6行を表示しています。各列の意味は以下の通りです:
- 行名(例:ENSMUSG00000069516): マウス遺伝子のEnsembl ID。ENSMUSGはMus musculus(マウス)の遺伝子を表し、一意の識別子として機能します。
- gene_name: 遺伝子の一般的な記号名(シンボル)。例えば「Lyz2」はリゾチーム2遺伝子を表します。
- gene_chr: 遺伝子が位置する染色体番号。マウスには19対の常染色体とX/Y性染色体があります。
- gene_start: 遺伝子の開始位置(塩基対単位)。例えば、Lyz2遺伝子は10番染色体の117,277,331番目の塩基から始まります。
- gene_end: 遺伝子の終了位置。Lyz2遺伝子は10番染色体の117,282,321番目の塩基で終わります。
- gene_strand: 遺伝子が位置するDNA鎖。「+」は正の鎖、「-」は負の鎖を示します。負の鎖にある遺伝子(Lyz2など)は逆向きに転写されます。
- gene_length: 遺伝子の長さ(塩基対単位)。例えば、Lyz2遺伝子は1,316塩基対の長さです。
- gene_biotype: 遺伝子の種類。ここでは全て「protein_coding」(タンパク質コード遺伝子)ですが、他にもncRNA(非コードRNA)などの種類があります。
- gene_description: 遺伝子の詳細な説明と出典情報。
- tf_family: 転写因子ファミリー。「-」は転写因子ではないことを示します。表示されている6つの遺伝子はいずれも転写因子ではありません。
表示されている6つの遺伝子はいずれも高発現のタンパク質コード遺伝子で、例えばLyz2(リゾチーム2)は抗菌活性を持つ酵素、Fth1(フェリチン重鎖)は鉄貯蔵タンパク質をコードしています。
> summary(gene_annotation) gene_name gene_chr gene_start gene_end Length:25529 Length:25529 Min. : 66 Min. : 1024 Class :character Class :character 1st Qu.: 36600485 1st Qu.: 36669013 Mode :character Mode :character Median : 74429500 Median : 74476068 Mean : 75782972 Mean : 75821730 3rd Qu.:109095365 3rd Qu.:109122895 Max. :195136811 Max. :195176716 gene_strand gene_length gene_biotype gene_description Length:25529 Min. : 38 Length:25529 Length:25529 Class :character 1st Qu.: 1235 Class :character Class :character Mode :character Median : 2816 Mode :character Mode :character Mean : 3755 3rd Qu.: 5105 Max. :123179 tf_family Length:25529 Class :character Mode :charactersummary(gene_annotation) の出力解説:
この出力は各列の統計的な概要を提供しています:
- gene_name, gene_chr, gene_strand, gene_biotype, gene_description, tf_family: これらは文字列データ(character)であるため、データフレーム内の要素数(Length)、データ型(Class)、保存モード(Mode)のみが表示されています。全て25,529個の要素を持ちます。
- gene_start:
- Min: 最小値は66、つまりゲノム上で最も5’側(開始位置が小さい)の遺伝子です
- 1st Qu.(第1四分位数): 36,600,485
- Median(中央値): 74,429,500
- Mean(平均値): 75,782,972
- 3rd Qu.(第3四分位数): 109,095,365
- Max(最大値): 195,136,811、最も3’側(終点が大きい)の遺伝子の開始位置
- gene_end:
- Min: 最小値は1,024、最も5’側の遺伝子の終了位置
- 1st Qu.~Max: それぞれ開始位置より大きな値になっています
- gene_length:
- Min: 最短の遺伝子は38塩基対で、これは非常に短い機能的RNAか、部分的な遺伝子注釈を示しています
- 1st Qu.: 25%の遺伝子は1,235塩基対以下
- Median: 中央値は2,816塩基対
- Mean: 平均は3,755塩基対で、中央値より大きいことから、一部の非常に長い遺伝子が平均を引き上げていることがわかります
- 3rd Qu.: 75%の遺伝子は5,105塩基対以下
- Max: 最長の遺伝子は123,179塩基対で、これは大規模な構造遺伝子や複雑なスプライシングを持つ遺伝子の可能性があります
遺伝子長の分布は右に歪んでおり(平均>中央値)、大多数は比較的短い(約1〜5kb)ですが、一部の非常に長い遺伝子があることがわかります。
# 遺伝子バイオタイプの分布を確認
biotype_counts <- table(gene_annotation$gene_biotype)
biotype_counts <- sort(biotype_counts, decreasing = TRUE)
head(biotype_counts, 10) # 上位10種類のバイオタイプを表示> head(biotype_counts, 10) protein_coding pseudogene lncRNA processed_pseudogene 19379 2024 1873 1321
transcribed_processed_pseudogene transcribed_unprocessed_pseudogene transcribed_unitary_pseudogene 372 339 77 unprocessed_pseudogene TEC snRNA 72 25 24head(biotype_counts, 10) の出力解説:
この出力は、データセット内の上位10種類の遺伝子バイオタイプとその遺伝子数を示しています:
| バイオタイプ | 遺伝子数 | 説明 |
|---|---|---|
| protein_coding | 19,379 | タンパク質をコードする遺伝子で、最も多い種類。リボソームでタンパク質に翻訳されるmRNAを生成します。 |
| pseudogene | 2,024 | 機能を失った遺伝子の複製。元々は機能的だったが、変異により翻訳能力を失っています。 |
| lncRNA | 1,873 | 長鎖非コードRNA。200塩基以上の長さがあり、タンパク質にコードされませんが、遺伝子発現調節など様々な機能を持ちます。 |
| processed_pseudogene | 1,321 | RNAが逆転写されてゲノムに再挿入されたことで生じた偽遺伝子で、イントロンを欠いています。 |
| transcribed_processed_pseudogene | 372 | 転写される処理済み偽遺伝子。機能的RNAを生成する可能性があります。 |
| transcribed_unprocessed_pseudogene | 339 | 転写される未処理の偽遺伝子。イントロンを保持しています。 |
| transcribed_unitary_pseudogene | 77 | 転写される単一性偽遺伝子。他の種では機能的だが、当該種では偽遺伝子化したもの。 |
| unprocessed_pseudogene | 72 | 未処理の偽遺伝子。遺伝子重複後に機能を失ったもので、イントロン構造を保持しています。 |
| TEC | 25 | To be Experimentally Confirmed。実験的確認が必要な転写証拠。 |
| snRNA | 24 | 小核RNA。核内で様々なRNA処理機能を持つ短い非コードRNA。 |
この分布から、データセットには多様な遺伝子タイプが含まれていることがわかり、タンパク質コード遺伝子が圧倒的多数(約76%)を占めていますが、様々な非コード遺伝子も含まれています。特に偽遺伝子と長鎖非コードRNAが多いことは注目に値します。
このコードでは、処理済みの遺伝子アノテーション情報を読み込み、その内容を確認します。gene_biotypeの分布を見ることで、データセット内のタンパク質コード遺伝子、非コードRNA、偽遺伝子などの割合を把握できます。
遺伝子の染色体分布の可視化
遺伝子がゲノム上でどのように分布しているかを確認します。
library(ggplot2)
# 染色体ごとの遺伝子数をカウント
chr_counts <- table(gene_annotation$gene_chr)
chr_df <- data.frame( chromosome = names(chr_counts), gene_count = as.numeric(chr_counts)
)
# 染色体番号で並べ替え(X, Y, MTを最後に)
chr_order <- c(as.character(1:19), "X", "Y", "MT")
chr_df$chromosome <- factor(chr_df$chromosome, levels = chr_order)
chr_df <- chr_df[order(chr_df$chromosome), ]
# 染色体分布のプロット
ggplot(chr_df, aes(x = chromosome, y = gene_count)) + geom_bar(stat = "identity", fill = "steelblue") + theme_minimal() + labs( title = "Number of Genes per Chromosome", x = "Chromosome", y = "Gene Count" ) + theme(axis.text.x = element_text(angle = 45, hjust = 1))このプロットでは、各染色体に位置する遺伝子の数を視覚化します。一般的に、より大きな染色体(例:染色体1)はより多くの遺伝子を持ちますが、遺伝子密度は染色体によって異なります
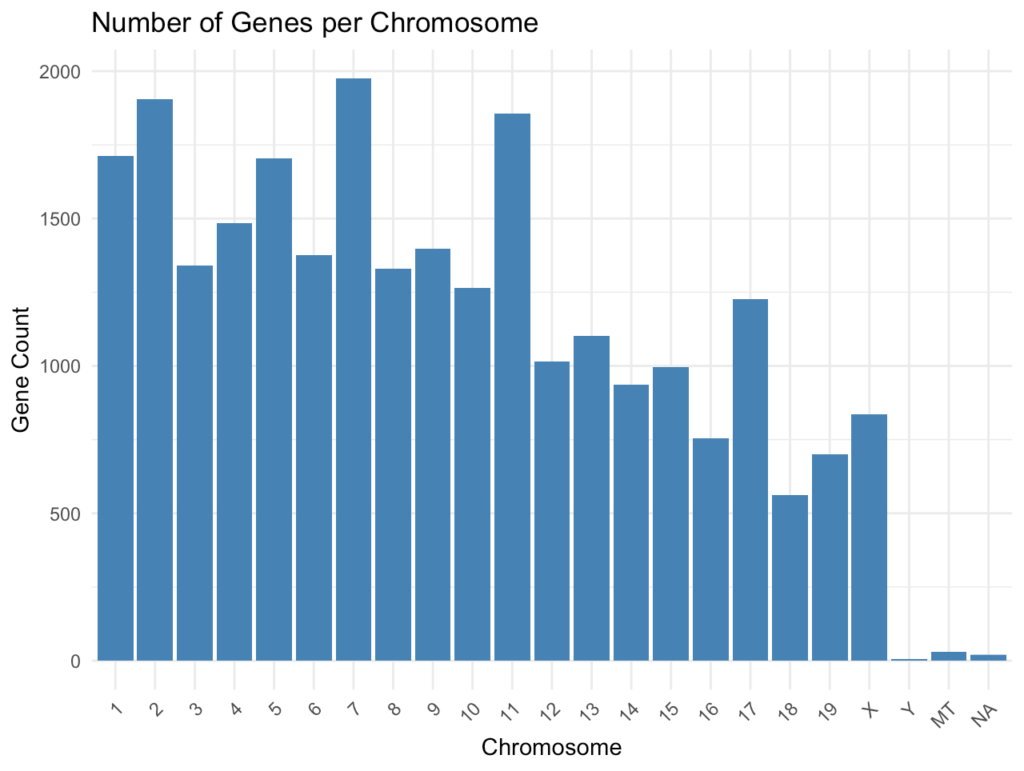
転写因子の抽出と分析
転写因子は遺伝子発現の調節に重要な役割を果たします。転写因子遺伝子を抽出して分析します。
# 転写因子を抽出
tf_genes <- gene_annotation[gene_annotation$tf_family != "-", ]
tf_family_counts <- table(tf_genes$tf_family)
tf_family_counts <- sort(tf_family_counts, decreasing = TRUE)
# 上位10個の転写因子ファミリーを表示
top_tfs <- head(tf_family_counts, 10)
tf_top_df <- data.frame( family = names(top_tfs), count = as.numeric(top_tfs)
)
# 転写因子ファミリーの分布をプロット
ggplot(tf_top_df, aes(x = reorder(family, -count), y = count)) + geom_bar(stat = "identity", fill = "darkred") + theme_minimal() + labs( title = "Top 10 Transcription Factor Families", x = "TF Family", y = "Count" ) + theme(axis.text.x = element_text(angle = 45, hjust = 1))このプロットでは、データセット内の主要な転写因子ファミリーとそれぞれの遺伝子数を示します。これにより、どの種類の転写因子が豊富に表現されているかがわかります。
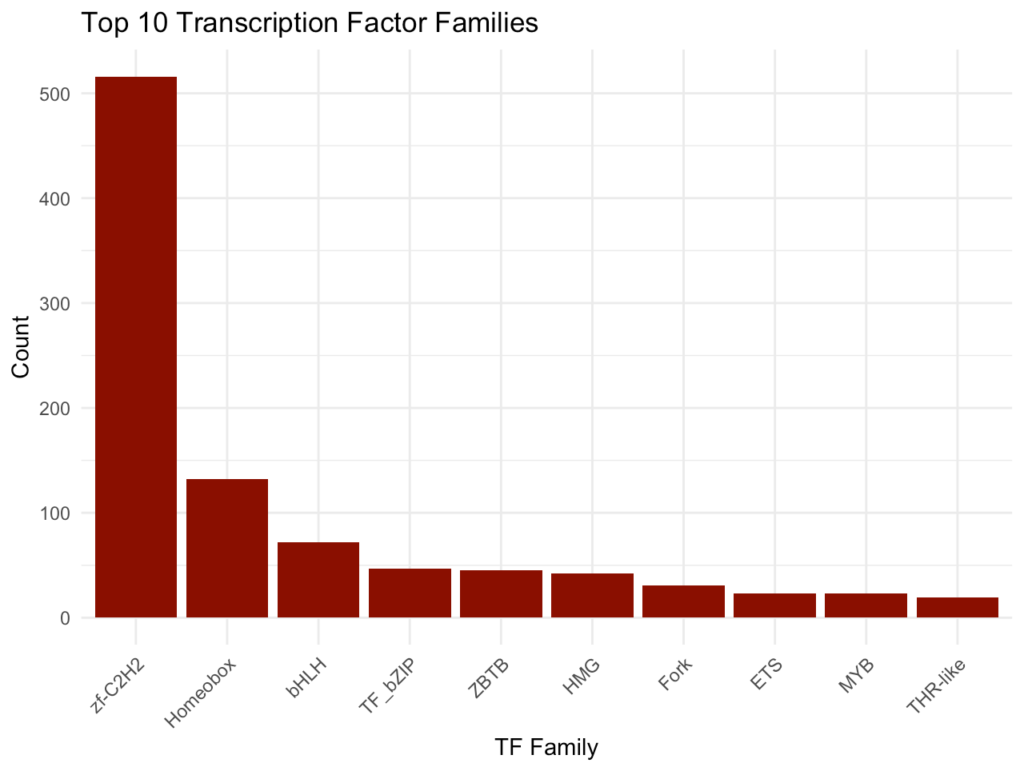
遺伝子長の分布確認
遺伝子の長さの分布を確認することで、短い遺伝子と長い遺伝子の割合を把握できます。
# 遺伝子長の分布をヒストグラムで表示
ggplot(gene_annotation, aes(x = gene_length)) + geom_histogram(bins = 50, fill = "skyblue", color = "black") + scale_x_log10() + # 遺伝子長は対数スケールで表示すると分布が見やすい theme_minimal() + labs( title = "Distribution of Gene Lengths", x = "Gene Length (log10 scale)", y = "Frequency" )
# 遺伝子バイオタイプ別の遺伝子長分布
# 上位5つのバイオタイプのみを抽出
top_biotypes <- names(head(biotype_counts, 5))
gene_subset <- gene_annotation[gene_annotation$gene_biotype %in% top_biotypes, ]
ggplot(gene_subset, aes(x = gene_length, fill = gene_biotype)) + geom_density(alpha = 0.5) + scale_x_log10() + theme_minimal() + labs( title = "Gene Length Distribution by Biotype", x = "Gene Length (log10 scale)", y = "Density", fill = "Gene Biotype" )最初のプロットは全遺伝子の長さの分布を、2番目のプロットは主要な遺伝子バイオタイプ別の長さ分布を示します。これにより、異なるタイプの遺伝子(例:タンパク質コード遺伝子vs長鎖非コードRNA)の長さの特性を比較できます。

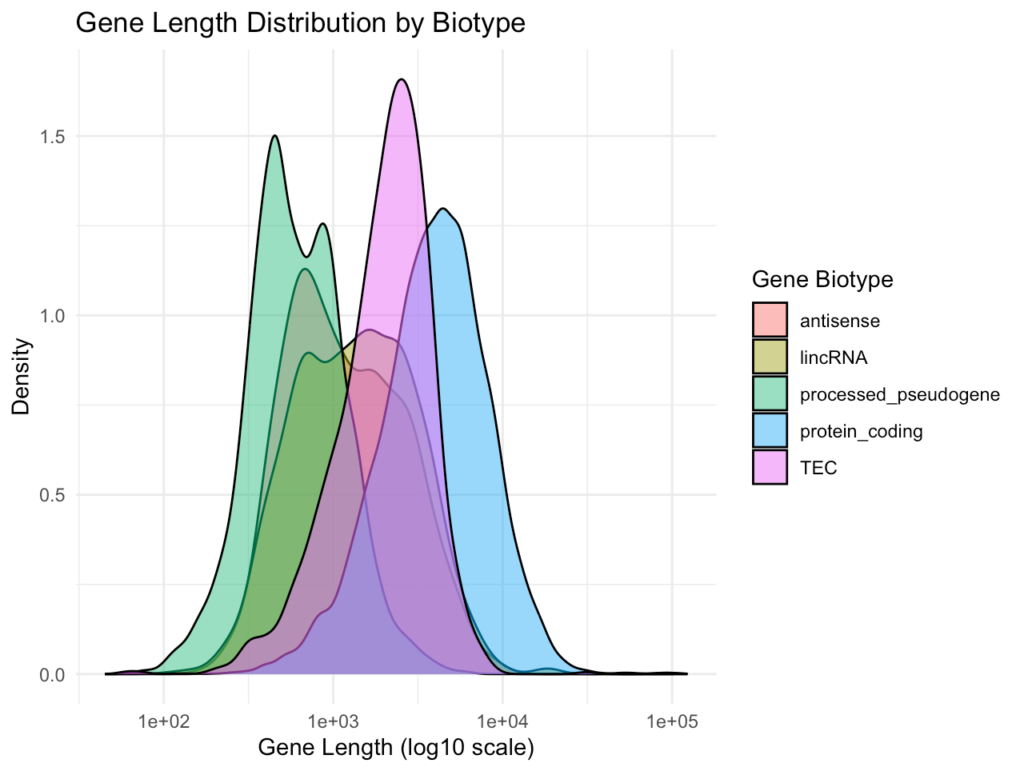
発現量データとの統合分析
遺伝子アノテーションと発現量データを組み合わせて、より深い分析が可能です。
# 正規化したカウントデータの読み込み
cpm_counts <- read.csv("data/processed/cpm_counts.csv", row.names = 1)
# 平均発現量の計算
mean_expr <- rowMeans(cpm_counts)
# 遺伝子アノテーションと発現量データの結合
gene_data <- cbind(gene_annotation, mean_expression = mean_expr)
# バイオタイプ別の平均発現量を可視化
# 上位5つのバイオタイプのみを抽出
top_biotypes <- names(head(biotype_counts, 5))
gene_subset <- gene_data[gene_data$gene_biotype %in% top_biotypes, ]
ggplot(gene_subset, aes(x = gene_biotype, y = mean_expression, fill = gene_biotype)) + geom_boxplot() + scale_y_log10() + theme_minimal() + theme( axis.text.x = element_text(angle = 45, hjust = 1), legend.position = "none" ) + labs( title = "Mean Expression by Gene Biotype", x = "Gene Biotype", y = "Mean Expression (log10 CPM)" )このプロットでは、異なる遺伝子タイプの平均発現レベルを比較しています。一般的に、タンパク質コード遺伝子は非コードRNAよりも高い発現レベルを示す傾向があります。
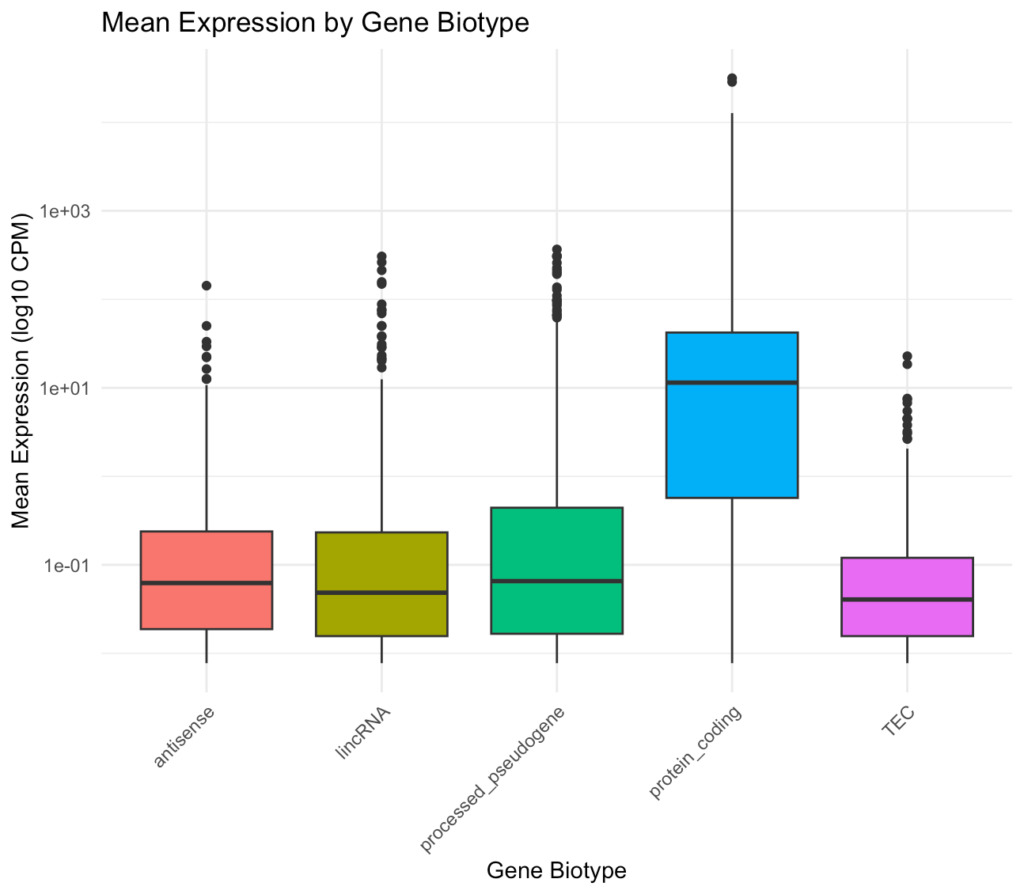
特定の遺伝子ファミリーや経路の抽出
特定の遺伝子ファミリーや関心のある経路に関連する遺伝子を抽出して分析することもできます。
# 例:キーワードで遺伝子を検索(サイトカイン関連遺伝子)
cytokine_genes <- gene_annotation[grep("cytokine|interleukin|chemokine", gene_annotation$gene_description, ignore.case = TRUE), ]
# 見つかったサイトカイン関連遺伝子の数
nrow(cytokine_genes)
# [1] 160
# サイトカイン関連遺伝子リストの表示
head(cytokine_genes[, c("gene_name", "gene_description")])
# gene_name
# ENSMUSG00000018927 Ccl6
# ENSMUSG00000026981 Il1rn
# ENSMUSG00000043953 Ccrl2
# ENSMUSG00000000982 Ccl3
# ENSMUSG00000003882 Il7r
# ENSMUSG00000027399 Il1a
# gene_description
# ENSMUSG00000018927 chemokine (C-C motif) ligand 6 [Source:MGI Symbol;Acc:MGI:98263]
# ENSMUSG00000026981 interleukin 1 receptor antagonist [Source:MGI Symbol;Acc:MGI:96547]
# ENSMUSG00000043953 chemokine (C-C motif) receptor-like 2 [Source:MGI Symbol;Acc:MGI:1920904]
# ENSMUSG00000000982 chemokine (C-C motif) ligand 3 [Source:MGI Symbol;Acc:MGI:98260]
# ENSMUSG00000003882 interleukin 7 receptor [Source:MGI Symbol;Acc:MGI:96562]
# ENSMUSG00000027399 interleukin 1 alpha [Source:MGI Symbol;Acc:MGI:96542]このコードでは、遺伝子の説明からキーワード検索を使用して、サイトカイン関連遺伝子を特定しています。これは、特定の生物学的プロセスや経路に関連する遺伝子セットを抽出したい場合に便利です。
遺伝子アノテーション情報を活用することで、RNA-seqデータの生物学的コンテキストをより深く理解できます。これは、差異発現分析や経路解析の結果を解釈する際に特に重要になります。
最後に
いかがだったでしょうか。
遺伝子アノテーション情報を活用することで、RNA-seq解析における解釈力が大きく向上します。ぜひご自身の解析にも取り入れて、より深い生物学的インサイトを得てみてください。
公共データを用いたSingle Cell RNA-seq解析に関する初心者向け技術書を販売中
プログラミング初心者でも始められるわかりやすい解説!
RとSeuratで始めるSingle Cell RNA-seq解析!
公共データを用いたシングルセル ダイナミクス解析に関する初心者向け技術書を販売中
シングルセルデータの高度な解析であるTrajectory解析、RNA Velocity解析、空間トランスクリプトーム解析が環境構築方法から詳しく解説されています!
TCGAデータ x R を用いた生存時間解析に関する技術書を販売中
R言語による生存時間解析の手法をステップバイステップで解説!自分のPCとRさえあれば、費用をかけずに本格的なサバイバル解析を体験できます

